
Врожденный буллезный эпидермолиз: Врожденный буллезный эпидермолиз

ВРОЖДЕННЫЙ БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ У ДЕТЕЙ: ВОПРОСЫ ЭТИОПАТОГЕНЕЗА, КЛИНИКИ, ДИАГНОСТИКИ И ЛЕЧЕНИЯ | Сомова
1. Кубанов А. А., Альбанова В. И., Карамова А. Э. и др. Распространенность врожденного буллезного эпидермолиза у населения Российской Федерации // Вестн. дерматологии и венерологии. 2015. № 3. С. 21–30.
2. The Epidermolysis Hereditary Bullous // Orphanet Consumer
3. Encyclopedia – First edition: Sept 2012. URL: www.orpha.net/data/patho/Pub/f /EpidermolyseBulleuseHereditaire-FRfrPub11387.pdf (дата обращения: 13.06.2020).
4. Снарская Е. С., Кряжева С. С., Карташова М. Г., Бобров М. А., Филатова И. В. Врожденный дистрофический гиперпластический буллезный эпидермолиз Коккейна – Турена // Рос. журн. кожных и венерич. болезней. 2011. № 5. С. 34–41.
журн. кожных и венерич. болезней. 2011. № 5. С. 34–41.
5. Fine J. D., Bruckner-Tuderman L., Eady R. A. et al. Inherited
6. Epidermolysis Bullosa: Updated Recommendations on Diagnosis
7. and Classification // J Am Acad Dermatol. 2014. № 6. Р. 1103–1126.
8. Коталевская Ю. Ю., Марычева Н. М. Буллезный эпидермолиз: основные клинические проявления // Педиатрия. № 4. 2014. С. 70–72.
9. Кубанова А. А., Мурашкин Н. Н. Особенности современного течения и эпидемиология буллезного эпидермолиза в Краснодарском крае // Вестн. дерматологии и венерологии. 2011. № 1. С. 59–65.
10. Fine J. Inherited Epidermolysis Bullosa // Orphanet J Rare Dis. 2010. № 5. P. 12. DOI 10.1186/1750-1172-5-12.
Fine J. Inherited Epidermolysis Bullosa // Orphanet J Rare Dis. 2010. № 5. P. 12. DOI 10.1186/1750-1172-5-12.
11. Fine J. D. Epidermolysis Bullosa Registries and the Epidemiology of Epidermolysis Bullosa (EB) // Blistering Diseases. 2015. № 1. P. 265–274. DOI 10.1007/978-3-662-45698-9_22.
12. Новиков П. В. Правовые аспекты редких (орфанных) заболеваний в России и в мире // Медицина. 2013. № 4. С. 50–70.
13. Федеральные клинические рекомендации по ведению больных врожденным буллезным эпидермолизом. М., 2015. 24 с.
14. Родин А. Ю., Сердюкова Е. А., Щава С. Н. Неинфекционные буллезные дерматозы. Волгоград : Изд-во ВолгГМУ, 2013. 132 с.
15. Kiritsi D., Pigors M., Tantcheva-Poor I., Wessel C., Arin M. J., Kohlhase J. et al. Epidermolysis Bullosa Simplex Ogna Revisited // J Invest Dermatol. 2013. № 133. P. 270–273.
Kiritsi D., Pigors M., Tantcheva-Poor I., Wessel C., Arin M. J., Kohlhase J. et al. Epidermolysis Bullosa Simplex Ogna Revisited // J Invest Dermatol. 2013. № 133. P. 270–273.
16. Yuen W. Y., Pasmooij A., Stellingsma C., Jonkman M. F. Enamel Defects in Carriers of a Novel LAMA3 Mutation Underlying Epidermolysis Bullosa // Acta Derm Venereol. 2012. № 92. Р. 695–696.
17. Fine J. D., Bruckner-Tuderman L., Eady R. A. J. et al. Inherited Epidermolysis Bullosa: Updated Recommendations on Diagnosis and Classification // J Am Acad Dermatol. 2014. Vol. 70. № 6. P. 1103–1126.
18. Wilson N. J., Messenger A. G., Leachman S. A., O’Toole E. A., Lane E. B., McLean W. H., Smith F. J. Keratin K6c Mutations Cause Focal Palmoplantar Keratoderma // J Invest Dermatol. 2010.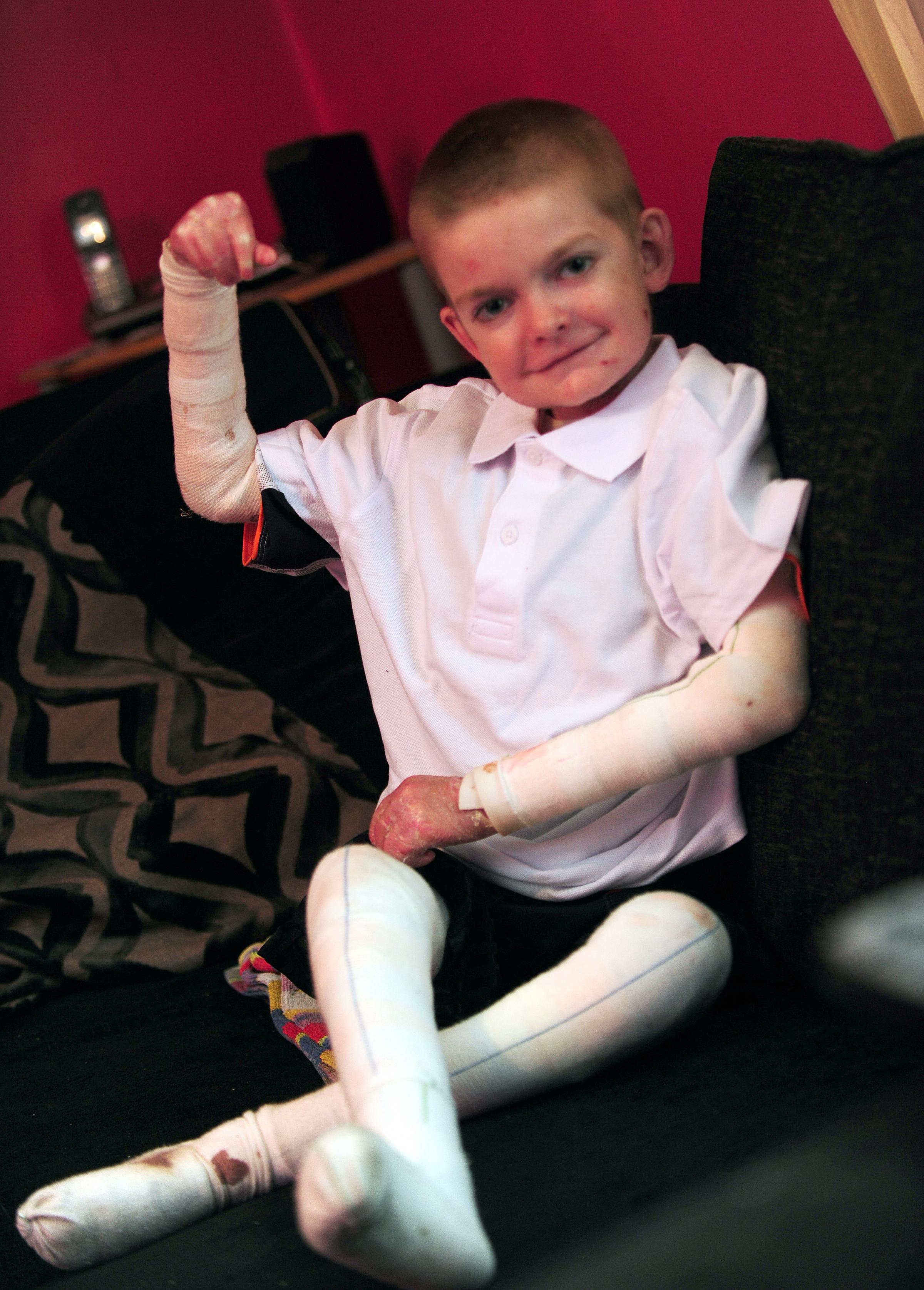 № 130. P. 425–429.
№ 130. P. 425–429.
19. Pigors M., Kiritsi D., Cobzaru C., Schwieger-Briel A., Suarez J., et al. TGM5mutations Impact Epidermal Differentiation in Acral Peeling Skin Syndrome // J Invest Dermatol. 2012. № 132. P. 2422–2429.
20. Pigors M., Schwieger-Briel A., Leppert J., Kiritsi D., Kohlhase J., Bruckner-Tuderman L. et al. Molecular Heterogeneity of
21. Epidermolysis Bullosa Simplex: Contribution of EXPH5 Mutations // J Invest Dermatol. 2014. №134. P. 842–845.
22. Yuen W. Y., Pas H. H., Sinke R. J., Jonkman M. F. Junctional
23. Epidermolysis Bullosa of Late Onset Explained by Mutations in
24. COL17A1 // Br J Dermatol. 2011. No. 164. P. 1280–1284.
COL17A1 // Br J Dermatol. 2011. No. 164. P. 1280–1284.
25. Natsuga K., Nishie W., Nishimura M. et al. Loss of Interaction
26. between Plectin and Type XVII Collagen Results in Epidermolysis Bullosa Simplex // Human Mutation. 2017. No. 9. P. 1666–1670. URL: https://onlinelibrary.wiley.com/doi/abs/10.1002/humu.23344 (дата обращения: 13.06.2020).
27. Kamaguchi M., Iwata H., Ujiie H., Natsuga K., Nishie W., Kitagawa Y., Shimizu H. High Expression of Collagen XVII Compensates for its Depletion Induced by Pemphigoid IgG in the Oral Mucosa // Journal of Investigative Dermatology. 2018. № 3. Р. 1707–1715. DOI 10.1016/j.jid.2018.03.002.
28. Альбанова В. И., Гольченко В. А. Лечение буллезного эпидермолиза // Рос. журн. кожных и венерич. болезней. 2013. № 4. С. 21–24.
журн. кожных и венерич. болезней. 2013. № 4. С. 21–24.
29. Сердюкова Е. А., Попов В. В. Лечение врожденного буллезного эпидермолиза у детей // Лекарств. вестн. 2016. Т. 10, № 4 (64). С. 43–47.
30. Goldschneider K. R., Good J., Harrop E. et al. Pain Care for Patients with Epidermolysis Bullosa: Best Care Practice Guidelines // BMC Medicine. 2014. № 12. P. 178.
31. Fujita Y., Abe R., Inokuma D., Sasaki M., Hoshina D., Natsuga K. et al. Bone Marrow Transplantation Restores Epidermal Basement Membrane Protein Expression and Rescues Epidermolysis Bullosa Model Mic // Proc Natl Acad Sci USA. 2010. № 107 (32). P. 14345–14350. DOI 10.1073/pnas.1000044107.
32. Tukaj S., Bieber K., Witte M. et al. Calcitriol Treatment Ameliorates Inflammation and Blistering in Mouse Models of Epidermolysis Bullosa Acquisita // Journal of Investigative Dermatology. 2018. Vol. 138, P. 301–309. DOI 10.1016/j.jid.2017.09.009.
et al. Calcitriol Treatment Ameliorates Inflammation and Blistering in Mouse Models of Epidermolysis Bullosa Acquisita // Journal of Investigative Dermatology. 2018. Vol. 138, P. 301–309. DOI 10.1016/j.jid.2017.09.009.
Что такое буллёзный эпидермолиз? | Университетская клиника г. Фрайбурга
Уход за новорожденными с подозрением на буллезный эпидермолиз
Основные правила
Максимально осторожное и нежное обращение с новорожденными и младенцами. Необходимо свести к минимуму любые травмирующие кожу факторы, т. к. это может спровоцировать возникновение пузырей. Нельзя использовать обычные перевязочные материалы. Скрининг новорожденного должен проводится путем осторожного забора крови из вены. При подозрении на наличие буллезного эпидермолиза следует как можно скорее провести диагностические исследования, включающие в себя биопсию кожи и анализ на мутации.
Общие рекомендации
Не использовать обычный пластырь или другие обычные самоклеящиеся повязки. Если они все же по ошибке были использованы, их следует очень аккуратно удалить с помощью специальных растворяющих средств (напр., Niltac).
Если они все же по ошибке были использованы, их следует очень аккуратно удалить с помощью специальных растворяющих средств (напр., Niltac).
При мониторинге или проведении ЭКГ не наклеивать электроды непосредственно на кожу, а осуществлять наблюдение с помощью пульсоксиметрии или фиксировать электроды с помощью силиконовых лент Mepilex transfer или Mepitac.
После купания не растирать кожу ребенка, а очень аккуратно обсушивать.
Ни в коем случае не поднимать и не носить ребенка, взяв под мышки. Чтобы поднять ребенка, следует сначала подложить под его тело пеленку, затем повернуть его на бок, подложить под пеленку руки таким образом, чтобы одна рука поддерживала голову и плечи, а вторая – нижнюю часть туловища ребенка. Поднимать ребенка как можно осторожнее, одним движением. Укладывать ребенка рекомендуется на ватный или мягкий матрас из пеноматериала, во время пребывания в клинике использовать специальный противопролежневый матрас из вискоэластика. При транспортировке следует выложить внутреннюю поверхность переносной колыбели мягким пеноматериалом (напр. Ligasano), чтобы предотвратить образование пузырей при усиленном потоотделении.
Ligasano), чтобы предотвратить образование пузырей при усиленном потоотделении.
Одежда ребенка должна быть свободной и мягкой, все предметы одежды должны одеваться швами наружу, предпочтение следует отдавать ползункам с интегрированным носком. Не одевать ребенка слишком тепло, это может вызвать усиленное потение и спровоцировать образование пузырей. Носочки должны быть без тугих резинок и не давить на кожу. Имеющиеся эластичные резинки следует отрезать. Обувь ребенка должна быть свободной и мягкой.
Кормление/питание
Для кормления и для успокоения ребенка можно использовать соски и пустышки Habermann. Если ребенок в состоянии брать грудь, возможно также и грудное вскармливание. В таком случае вокруг рта и на щеки ребенка, а также вокруг соска матери необходимо нанести вазелин или мазь бепантен, что позволяет минимизировать трение кожи.
Если ребенок не голоден, однако ему требуется успокоительное сосание, можно предложить ему успокоительную соску-пустышку. Для этого рекомендуется использовать соски из натурального латекса с мягкой эластичной защитной пластинкой.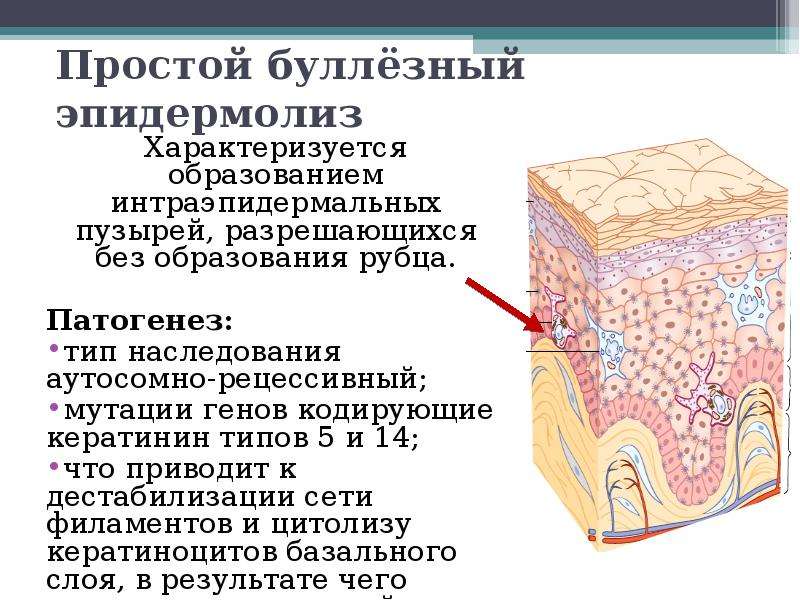 Тем не менее, надо иметь в виду, что сосание пустышки также может способствовать образованию пузырей. Начиная с 10-го дня жизни требуется проводить заместительную терапию витамином Д, а также препаратами фтора. Прием фтора необходим в связи с тем, что гигиена полости рта у пациентов с БЭ может быть сильно затруднена.
Тем не менее, надо иметь в виду, что сосание пустышки также может способствовать образованию пузырей. Начиная с 10-го дня жизни требуется проводить заместительную терапию витамином Д, а также препаратами фтора. Прием фтора необходим в связи с тем, что гигиена полости рта у пациентов с БЭ может быть сильно затруднена.
Требуется особенно тщательно наблюдать за ростом и развитием ребенка с заболеванием БЭ, т.к. постоянные процессы заживления кожи требуют повышенного расхода калорий. Детей нужно регулярно взвешивать. При недостаточном развитии ребенка по согласованию с педиатром необходимо дополнительно к материнскому молоку ввести в его рацион специальные питательные добавки (напр. Maltodextrin).
Частым осложнением БЭ являются запоры. Если достаточное количество жидкости и специальное питание не способствуют смягчению консистенции стула, то назначаются не свечи, а оральные препараты (напр. Lactulose).
Общие правила ухода за кожей, пупком и руками
В целом, необходимости в специальном уходе за здоровыми участками кожи младенцев нет. Важно обрабатывать корочки, образующиеся в процессе заживления пузырей, смягчающими мазями (напр. Linola, Bepanthen) и затем аккуратно удалять их, т.к. в противном случае они могут вызывать раздражение кожи и возникновенение новых пузырьков.
Важно обрабатывать корочки, образующиеся в процессе заживления пузырей, смягчающими мазями (напр. Linola, Bepanthen) и затем аккуратно удалять их, т.к. в противном случае они могут вызывать раздражение кожи и возникновенение новых пузырьков.
До отпадения пуповины с целью предотвращения трения и возникновения пузырей необходимо непосредственно вокруг пупка накладывать силиконовую губчатую повязку (напр. Mepilex lite или transfer).
Ногти младенцам следует обрезать только тогда, когда они станут очень длинными и появится опасность, что ребенок сможет поранить себя. Следует учитывать то, что сам процесс обрезания также может вызвать образование пузырей, поэтому по возможности делать это следует во время сна ребенка.
Если ребенок сосет пальцы, то на ручки надо одевать специальные варежки или носочки.
Обработка пузырей и ран
Пузыри следует прокалывать большой стерильной канюлей в нескольких местах, за 2 минуты до этого детям грудного возраста с целью обезболивания следует дать 1 – 2 мл G40% орально. При уходе за эрозиями и пузырями нельзя использовать сухие повязки, т.к. они могут приклеиваться и в дальнейшем при замене повреждать кожу и провоцировать возникновение новых пузырей.
При уходе за эрозиями и пузырями нельзя использовать сухие повязки, т.к. они могут приклеиваться и в дальнейшем при замене повреждать кожу и провоцировать возникновение новых пузырей.
При инфицированных, гнойных пузырях, покрывающую их кожу необходимо удалить и продезинфицировать рану.
На свежие раны рекомендуется накладывать сетчатую накладку Urgotül или Mepitel, затем внешнюю повязку из силиконового губчатого материала (Mepilex lite, Mepilex transfer) и далее фиксировать все мягкой повязкой (напр. Medicomp), марлевым бинтом или трубчатой фиксирующей повязкой. Если имеется необходимость наклеить пластырь непосредственно на кожу, можно использовать повязку Mepitac.
При неинфицированных ранах, чтобы максимально оградить ребенка от болей, вызываемых процессом перевязки, мепитекс можно оставить на несколько дней (макс. 7 дней). Внешнюю повязку требуется менять каждый день.
При сухих открытых ранах для обеспечения увлажнения на или под сетчатую накладку можно наносить мази Bepanten или Prontosan.
Для минимизации травмирования кожи и предотвращения образования пузырей в местах преимущественной локализации (в области ягодиц, пяток и резинок подгузников) рекомендуется использовать повязки Mepilex, Mepilex lite и transfer.
При подозрении на инфицирование раны при каждой смене повязки ее следует в течение 5 минут обработать дезинфицирующим раствором (напр. Octenisept, Prontosan, Lavanid). Если это не приведет к улучшению состояния раны или появится жар, необходимо взять мазок и провести оральную антибиотическую терапию (напр. препаратом Cefalosporin 2-го поколения).
От повязок с содержанием серебра из-за опасности возникновения аргирии в первые месяцы жизни ребенка следует отказаться.
Если новорожденный содержится в инкубаторе, необходимо обязательно следить за тем, чтобы в нем не застаивался слишком теплый воздух, т.к. это увеличивает риск образования пузырей.
Обработка пузырей в области подгузников
Необходимо стараться максимально избегать трения в области подгузников, а также сильно не растирать кожу при ее обработке и очищении. Труднодоступные для очищения участки необходимо покрыть марлевой повязкой, пропитанной слоем Bepanten, и затем использовать ее для очищения при смене подгузников.
Труднодоступные для очищения участки необходимо покрыть марлевой повязкой, пропитанной слоем Bepanten, и затем использовать ее для очищения при смене подгузников.
Если у детей в области подгузников образовались пузыри, необходимо накладывать силиконовые повязки (Mepilex transfer). Эластичные резинки от подгузников рекомендуется срезать.
Перианальные трещины смазывать цинковой пастой. В остальном, применяются вышеуказанные общие правила обработки ран и ухода за кожей.
причины, симптомы, лечение, профилактика — клиника «Добробут»
Буллезный эпидермолиз: причины, симптомы, лечение
Буллезный эпидермолиз – это не одна патология, а целая группа наследственных нозологий, которые манифестируются легкой ранимостью кожных покровов. Основное проявление – формирование на коже пузырей с жидким содержимым, а после их вскрытия – образование эрозий, которые долго не заживают.
Причины и формы буллезного эпидермолиза
Причины буллезного эпидермолиза зависят от типа.
Простой буллезный эпидермолиз развивается из-за мутаций генов KRT5 и KRT14. Предполагается, что при этом в кожных покровах нарушается равновесие между ферментами и ингибиторами (соединениями, способными подавлять процессы в тканях). Как результат, выделяются ферменты, которые разрушают белки кожи, на фоне чего образуются пузыри.
Пограничный буллезный эпидермолиз провоцируют мутации в генах под маркировкой LAMB3 и LAMA3. Курируемая ими ферментная система становится разбалансированной, из-за чего страдают коллаген 17-го типа и ламинин-332, без которых нарушается нормальное строение кожи. Помимо формирования пузырей и эрозий, появляется усиленная ломкость кожи.
Дистрофический буллезный эпидермолиз развивается из-за мутаций в гене COL7A1. Из-за этого страдает коллаген 7-го типа, контролирующий состояние соединительнотканных элементов кожи. Нехватка этого протеина провоцирует образование на коже сыпи, эрозий (язвочек) и пузырей.
Особенностью смешанного буллезного эпидермолиза является формирование пузырей во всех слоях кожи.
Это основные типы патологии. На данный момент различают десятки видов буллезного эпидермолиза.
Симптомы
Симптомы патологии могут отличаться, но общий признак разных форм буллезного эпидермолиза – образование пузырей и язвочек при механическом воздействии на кожу.
При локализованной форме простого буллезного эпидермолиза изменения кожи наблюдаются только на одном участке тела (кисти, стопе).
Пограничная форма буллезного эпидермолиза характеризуется более тяжелым состоянием. Например, при летальном подтипе Херлитца диагностируются:
- усиленная ломкость кожи;
- формирование большого количества пузырей и эрозий;
- образование грануляций на лице и спине.
Нередко больные буллезным эпидермолизом летального подтипа умирают в первые годы жизни. У выживших наблюдаются:
- контрактуры (тугоподвижность) суставов;
- поражение почек;
- потеря ногтевых пластин.

Для атрофической формы пограничного буллезного эпидермолиза характерны обширные высыпания с образованием рубцов.
Дистрофический буллезный эпидермолиз в основном поражает большие участки тела. Его доминантный вариант (связанный с доминантными генами) более доброкачественный – такие больные теряют ногти, у них образуются заметные рубцы. Рецессивный вариант более тяжелый: при нем поражаются кости, а на месте шрамов с годами может возникнуть плоскоклеточный рак.
Диагностика
Диагноз буллезного эпидермолиза у детей и взрослых ставят на основании осмотра кожи, проведения иммуногистологических исследований и генетического анализа, наследственного анамнеза.
Главный тест заключается в том, что врач механически воздействует на кожу пациента и через некоторое время оценивает последствия такого раздражения.
При иммунофлуоресцентном анализе используют антитела, которые имеют сродство к белкам кожи. При помощи такого метода можно оценить количество белков – а значит, и ферментную активность тканей. Сниженный уровень белка подтверждает его низкое выделение либо форсированное разрушение.
Сниженный уровень белка подтверждает его низкое выделение либо форсированное разрушение.
Изучение наследственного анамнеза помогает выявить у пациента родственников с таким же заболеванием.
Осложнения
Важно знать о причинах буллезного эпидермолиза, а также возможных осложнениях. Буллезный эпидермолиз независимо от вида чаще всего сопровождается такими осложнениями:
- присоединение вторичной инфекции;
- инфекционно-токсический шок;
- сепсис;
- обезвоживание.
Буллезный эпидермолиз: лечение
Специфическое лечение не разработано. Цель терапевтических процедур – уменьшение выраженности кожных нарушений и предупреждение формирования осложнений.
При развитии тяжелых форм патологии назначают преднизолон.
Местное лечение заключается во вскрытии пузырьков, их обработке антисептиками. Повязку накладывают очень осторожно – давление способно спровоцировать развитие новых пузырей.
Также разрабатывают методы лечения с применением стволовых клеток, белковую и генную терапии – но пока они апробируются только на животных. На данный момент буллезный эпидермолиз является неизлечимой патологией.
Профилактика
Заболевание является врожденным, поэтому специфической профилактики нет. Для предупреждения развития проблем с кожей на фоне такой патологии следует очень бережно относиться к собственным кожным покровам – избегать травматизации.
Читайте детальнее о лечении буллезного эпидермолиза на нашем сайте Добробут.ком.
Врожденный буллезный эпидермолиз в практике врача-дерматовенеролога Текст научной статьи по специальности «Клиническая медицина»
УДК 616.5291 Клинический случай
ВРОЖДЕННЫЙ БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ В ПРАКТИКЕ ВРАЧА-ДЕРМАТОВЕНЕРОЛОГА
О.А. Шустова — ГУЗ «Саратовский областной кожно-венерологический диспансер», врач-дерматовенеролог: Н. К. Бобко — ГУЗ «(Саратовский областной кожно-венерологический диспансер», врач-дерматовенеролог.
К. Бобко — ГУЗ «(Саратовский областной кожно-венерологический диспансер», врач-дерматовенеролог.
EPIDERMOLYSIS BULLOSA IN PRACTICE OF DERMATOLOGIST
O.A. Shustova — Saratov Regional Dermatovenerologic Dispensary, Dermatologist; N. K. Bobko — Saratov Regional Dermatovenerologic Dispensary, Dermatologist.
Дата поступления — 4.09.2014 г Дата принятия в печать — 22.09.2014 г.
Шустова О. А., Бобко Н. К. Врожденный буллезный эпидермолиз в практике врача-дерматовенеролога. Саратовский научно-медицинский журнал 2014; 10 (3): 553-555.
В статье описан клинический случай редкого дерматоза — врожденного дистрофического буллезного эпи-дермолиза у ребенка 9 лет.
Ключевые слова: буллезный эпидермолиз, клиника.
Shustova OA, Bobko NK. Epidermolysis bullosa in practice of dermatologist. Saratov Journal of Medical Scientific Research 2014; 10 (3): 553-555.
This article describes a clinical case of a rare dermatosis — congenital dystrophic epidermolysis bullosa in a 9-year old child.
Key words: epidermolysis bullosa, clinical picture.
Введение. Буллезный эпидермолиз — это группа редких наследственных заболеваний, включающая около 30 форм, характеризующихся нарушением межклеточных контактов в эпидермисе или дерме, что при малейшей травме приводит к образованию пузырей [1, 2]
С учетом клинических особенностей и методов электронной микроскопии выделяют три основные группы врожденного буллезного эпидермолиза: простой буллезный эпидермолиз, пограничный буллезный эпидермолиз и дистрофический буллезный эпидермолиз [3].
При простой форме буллезного эпидермолиза причиной возникновения внутриэпидермальных пузырей служит мутация генов, кодирующих синтез кератина 5, 14 типов и плектина. Она приводит к дестабилизации сети тонофиламентов и цитолизу кератиноцитов базального слоя, в результате чего базальный слой отслаивается, при этом неповрежденная базальная мембрана находится в основании пузыря [4, 5].
Наследуются чаще всего по аутосомно-доминант-ному типу [4].
Простой буллезный эпидермолиз начинается с рождения или в первые дни жизни. Заболевание характеризуется появлением пузырей на местах механической травмы (локти, колени, кисти, стопы, поясница). Пузыри имеют различные размеры, прозрачное, редко — геморрагическое содержимое. Эрозии, образующиеся после вскрытия пузырей, быстро эпителизируются, не оставляя следов. Слизистые оболочки поражаются очень редко. В патологический процесс ногтевые пластинки обычно не вовлекаются [1, 2].
В патологический процесс ногтевые пластинки обычно не вовлекаются [1, 2].
При пограничной форме буллезного эпидермо-лиза пузыри образуются в результате расщепления светлой пластинки базальной мембраны на границе эпидермиса и дермы. Генетический дефект — мутации генов, кодирующих антиген буллезного пемфи-гоида (в гемидесмосомах) и ламинин 332 (в якорных филаментах). Плотная пластинка базальной мем-
Ответственный автор — Шустова Ольга Александровна
Тел.: 79372405057
E-mail: [email protected]
браны находится в основании пузыря. Тип наследования аутосомно-рецессивный [2, 4, 5].
К типичным симптомам данной формы относятся образование множества пузырей, эрозий и атрофи-ческих рубцов кожи, ониходистрофия, приводящая к полной утрате ногтевых пластин, тяжелое поражение мягких тканей в ротовой полости, гипоплазия эмали и тяжелый кариес.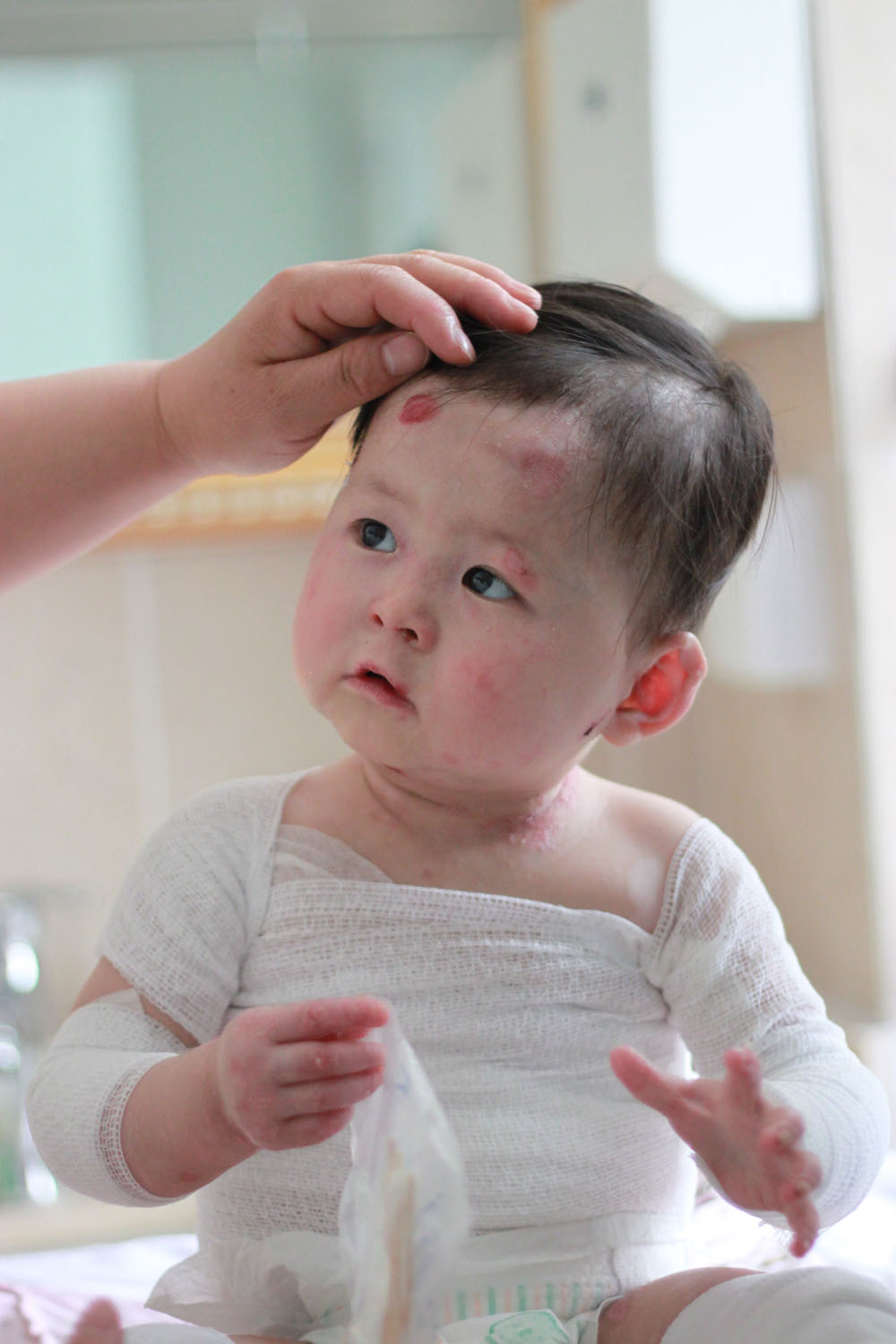 Патогномоничным симптомом является обильная грануляционная ткань, симметрично образующаяся вокруг рта, в области средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжелая полиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта, поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза [6].
Патогномоничным симптомом является обильная грануляционная ткань, симметрично образующаяся вокруг рта, в области средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжелая полиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта, поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза [6].
При дистрофическом буллезном эпидермолизе пузыри формируются глубоко под базальной мембраной, поэтому после заживления остаются рубцы. Развитие данной формы обусловлено мутацией гена, кодирующего коллаген VII типа — компонент крепящихся фибрилл. Из-за этого нарушения крепящиеся фибриллы рудиментарны или отсутствуют [2, 4, 5].
Описаны как аутосомно-рецессивные, так и ау-тосомно-доминантные варианты наследования дистрофического буллезного эпидермолиза [4].
Доминантный дистрофический буллезный эпидермолиз начинается с рождения или первых дней жизни. В первые месяцы поражение кожи генерализованное, в дальнейшем пузыри возникают обычно на одних и тех же часто травмируемых участках: кистях, стопах, коленях, локтях, шее. Заживление происходит с образованием атрофического рубца. Ногтевые пластинки поражены у всех больных, и лишь в редких случаях ногти отсутствуют, чаще они дистро-фичны. Рост и развитие детей не нарушены. С возрастом пузыри появляются все реже, и у взрослых о наличии болезни могут напоминать только дистрофические изменения ногтей и едва заметные рубцы на локтях, коленях и лодыжках [3, 7].
Рецессивный дистрофический буллезный эпидермолиз протекает тяжело, часто приводит к смерти в раннем возрасте. Заболевание всегда возникает с
554
ДЕРМАТОВЕНЕРОЛОГИЯ
рождения или первых часов жизни.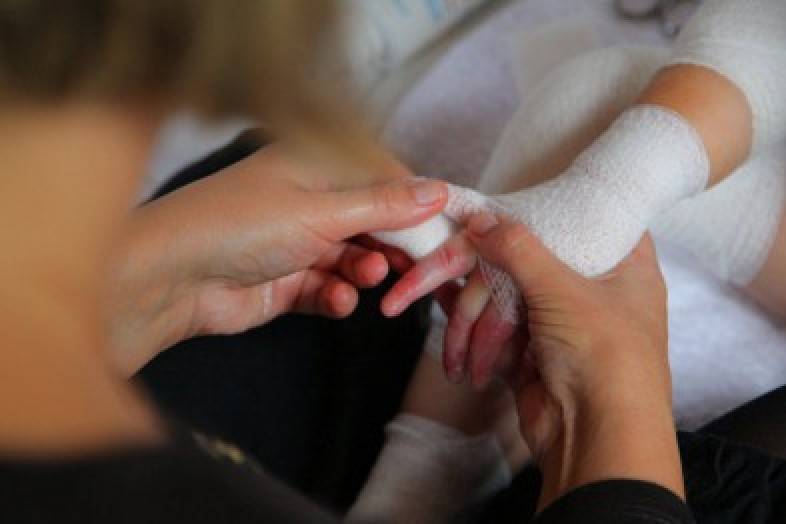 Уже при рождении часто эрозирована кожа конечностей.
Уже при рождении часто эрозирована кожа конечностей.
В первые дни жизни происходит распространение высыпаний. Заживление происходит с образованием атрофических рубцов, на кистях и стопах постепенно развиваются контрактуры и синдактилии. Ногтевые пластинки отсутствуют с рождения или постепенно утрачиваются в результате образования подногтевых пузырей. На слизистой оболочке полости рта, пищевода, прямой кишки также возникают множественные пузыри. Процесс рубцевания во рту приводит к ограничению подвижности языка, атрофии его сосочков, заращению вестибулярных складок и микростомии, в пищеводе — к его сужению, нарушению проходимости пищи, в прямой кишке — к хроническим запорам, резким болям при дефекации. Зубы поражены у всех больных, преобладают кариес, дефекты зубной эмали [1, 3, 6].
Этиотропного лечения буллезного эпидермолиза пока нет Поэтому лечение больных является симптоматическим. Выбор симптоматических методов зависит от тяжести и обширности поражения. Образование пузырей может быть минимизировано ограничением травматических воздействий и использованием мягкой, хорошо подобранной обуви, одежды [2, 4, 7].
Выбор симптоматических методов зависит от тяжести и обширности поражения. Образование пузырей может быть минимизировано ограничением травматических воздействий и использованием мягкой, хорошо подобранной обуви, одежды [2, 4, 7].
Описание клинического случая. Пациент А., 9 лет, состоит на диспансерном учете в ГУЗ « СОКВД» с 2005 г с диагнозом: «Врожденный буллезный эпи-дермолиз».
Родился от пятой беременности, вторые срочные роды. В родах 2-кратное обвитие пуповины. Масса тела при рождении 3 кг 450 г, рост 52 см. С рождения состояние тяжелое за счет поражения кожи. Кожный процесс носил распространенный характер: на коже туловища, верхних, нижних конечностей имелись множественные пузыри с серозно-геморрагическим содержимым, склонные к периферическому росту и слиянию. На месте вскрывшихся пузырей формировались обширные эрозии, с сочным ярко-красным дном. С рождения укорочение правой нижней конечности, ониходистрофия. В тяжелом состоянии переведен в отделение реанимации новорожденных, где было выполнено гистологическое исследование кожи, на основании которого выставлен диагноз: «Пограничный буллезный эпидермолиз тип Херлит-ца». По мере роста ребенка пузыри появлялись в основном в местах, подверженных механическому воздействию (голени, колени, предплечья, локти), а также могли возникнуть на любом участке кожного покрова (рис. 1).
В тяжелом состоянии переведен в отделение реанимации новорожденных, где было выполнено гистологическое исследование кожи, на основании которого выставлен диагноз: «Пограничный буллезный эпидермолиз тип Херлит-ца». По мере роста ребенка пузыри появлялись в основном в местах, подверженных механическому воздействию (голени, колени, предплечья, локти), а также могли возникнуть на любом участке кожного покрова (рис. 1).
С двухлетнего возраста отмечалось некоторое улучшение кожного процесса, высыпания в основном локализуются на шее, паховых областях.
С 200б г. состоит на диспансерном учете по месту жительства у гастроэнтеролога с диагнозом: «Сужение верхней трети пищевода, реактивные изменения поджелудочной железы, колодискинезия по гипомо-торному типу»; у ортопеда с диагнозом: «Гипоплазия костей правой стопы, варусная установка правой стопы, вальгусная установка левой стопы».
В 2013 г.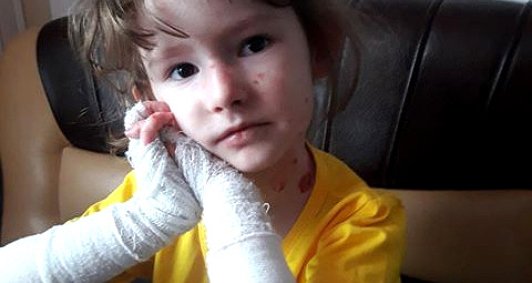 в Чешской Республике, в Университетской больнице г. Брно, исследовали биоптат кожи ребенка (методом антигенного картирования), а также был произведен ДНК-молекулярный анализ.
в Чешской Республике, в Университетской больнице г. Брно, исследовали биоптат кожи ребенка (методом антигенного картирования), а также был произведен ДНК-молекулярный анализ.
Антигенное картирование: белки кератин 5, кератин 14, коллаген XVII, ламинин 332: окрашиваются обычно, коллаген VII типа: окрашивается слабо.
Рис. 1. Пациент А, 4 мес.
м jp»
Рис.2. Пациент А., 9 лет
ДНК-молекулярный анализ: у пациента обнаружены 2 мутации в гене, отвечающие за синтез коллагена VII типа.
На основании анамнеза, клинической картины, лабораторных данных ребенку был выставлен диагноз: «Дистрофический буллезный эпидермолиз, ау-тосомно-рецессивный тип».
На сегодняшний день пузыри в основном локализуются в паховых областях, ладонной поверхности кистей.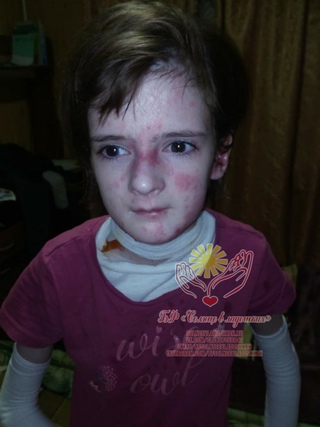 Кожный процесс представлен очагами эритемы ярко-розового цвета, с неровными, нечеткими контурами, на поверхности эрозии, с обрывками эпидермиса по периферии, местами покрытые серозно-геморрагическими корочками. (рис. 2). На коже груди, живота, верхних, нижних конечностях множественные атрофические рубцы, гиперпигментация.
Кожный процесс представлен очагами эритемы ярко-розового цвета, с неровными, нечеткими контурами, на поверхности эрозии, с обрывками эпидермиса по периферии, местами покрытые серозно-геморрагическими корочками. (рис. 2). На коже груди, живота, верхних, нижних конечностях множественные атрофические рубцы, гиперпигментация.
Ногтевые пластинки на правой руке отсутствуют на I и III пальцах, на левой руке на III пальце. На нижних конечностях ногтевые пластинки отсутствуют полностью.
За время диспансерного наблюдения пациент получает симптоматическую терапию, направленную на
Саратовский научно-медицинский журнал. 2014. Т. 10, № 3, дерматовенерология
предупреждение инфекционных осложнений со стороны кожи и стимуляцию репаративных процессов.
Заключение. Приведенное клиническое наблюдение интересно из-за редкой встречаемости данного дерматоза, трудности диагностики и отсутствия эффективных методов лечения. К сожалению, бул-лезный эпидермолиз — неизлечимое заболевание, однако это совсем не означает, что таким пациентам нельзя помочь. Основным в лечении болезни является правильный уход за кожей, который позволяет минимизировать осложнения и адаптировать таких людей в обществе. Необходимо отметить, что диспансерное наблюдение пациентов с данным заболеванием должно осуществляться в течение всей жизни.
Приведенное клиническое наблюдение интересно из-за редкой встречаемости данного дерматоза, трудности диагностики и отсутствия эффективных методов лечения. К сожалению, бул-лезный эпидермолиз — неизлечимое заболевание, однако это совсем не означает, что таким пациентам нельзя помочь. Основным в лечении болезни является правильный уход за кожей, который позволяет минимизировать осложнения и адаптировать таких людей в обществе. Необходимо отметить, что диспансерное наблюдение пациентов с данным заболеванием должно осуществляться в течение всей жизни.
Конфликт интересов отсутствует.
References (Литература)
1. Albanova VI. Hereditary pemphigus (epidermolysis bullosa). Russian Medical Journal 1997; 5 (11): 735-744. Russian (Альбанова В. И. Наследственная пузырчатка (бул-лезный эпидермолиз). Русский медицинский журнал 1997; 5 (11): 735-744. )
)
2. Wolf K, Johnson R, Syurmond D. Dermatology: Guidebook. M.: Practice, 2007; p. 138-153. Russian (Вульф К, Джонсон Р, Сюрмонд Д. Дерматология: Атлас-справочник. М.: Практика, 2007; с. 138-153)
3. Rodionov VG, Provision LN. Dystrophic epidermolysis bullosa (clinical case). Prospects of medicine and biology 2012; 4 (1): 91-94. Russian (Родионов В. Г., Провизион Л. Н. Дистрофический буллезный эпидермолиз (клиническое наблюдение). Перспективы медицины и биологии 2012; 4 (1): 91-94.)
4. Kay Shou-Mei Kane, Peter A. Lio. Pediatric Dermatology: Color Atlas and Reference. M.: Beanom, 2011; p. 94-104. Russian (Кей Шу-Мей Кейн, Питер А. Лио. Детская дерматология: Цветной атлас и справочник. М.: Бином, 2011; c. 94-104)
5. Albanova VI, Golchenko VA. Hereditary epidermolysis bullosa: current understanding of the etiology and pathogenesis. Russian Journal of Skin and Sexually Transmitted Diseases 2013; (2): 15-19. Russian (Альбанова В. И., Гольченко В. А. Наследственный буллезный эпидермолиз: современные представления об этиологии и патогенезе. Российский журнал кожных и венерических болезней 2013; (2): 15-19.)
Russian Journal of Skin and Sexually Transmitted Diseases 2013; (2): 15-19. Russian (Альбанова В. И., Гольченко В. А. Наследственный буллезный эпидермолиз: современные представления об этиологии и патогенезе. Российский журнал кожных и венерических болезней 2013; (2): 15-19.)
6. Mahneva NV, Andreeva TE. Cases of severe generalized recessive dystrophic epidermolysis bullosa. Russian Journal of Skin and Sexually Transmitted Diseases 2012; (3): 12-17. Russian (Махнева Н. В., Андреева Т. Е. Случай тяжелого генерализованного рецессивно-дистрофического врожденного буллезного эпидермолиза. Российский журнал кожных и венерических болезней 2012; (3): 12-17.)
7. Kaljuzhnaja LD. Clinical management of patients with epidermolysis bullosa. Ukrainian Journal of Dermatology, Venereology, Cosmetology 2003, (4): 27-29. Russian (Калюж-ная Л. Д. Тактика ведения больных с буллезным эпидермоли-зом. Украинский журнал дерматологии, венерологии, косметологии 2003; (4): 27-29. )
)
УДК [616.5-006.6-091.8:616.21] -085.831-036.8 (045) Краткое сообщение
РЕЗУЛЬТАТЫ ЛЕЧЕНИЯ РАЗЛИЧНЫХ МОРФОЛОГИЧЕСКИХ ТИПОВ РАКА КОЖИ ЛОР-ОРГАНОВ МЕТОДОМ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ
В. Н. Волгин — ФГКУ «Главный военный клинический госпиталь им. акад. Н. Н. Бурденко» (г. Москва), врач-дерматовенеролог, доктор медицинских наук; Е. Ф. Странадко — ФГБУ «Главный научный центр лазерной медицины Федерального медико-биологического агентства России» (г. Москва), профессор, доктор медицинских наук; Р. В. Каго-янц— ГБУЗ «Армавирский онкологический диспансер» (г. Армавир), врач-онколог.
THE RESULTS OF THE TREATMENT OF VARIOUS MORPHOLOGICAL TYPES OF ENT SKIN
CANCER BY PHOTODYNAMYC THERAPY
V. N. Volgin — Main Military Clinical Hospital n.a. Acad. N. N. Burdenko, Moscow, Dermatovenerologist, Doctor of Medical Sciences; E. F. Stranadko — Main Research Center of Laser Medicine, Federal medical-biological Agency of Russia, Moscow, Professor, Doctor of Medical Sciences, R. V. Kagoyants — Armavir Oncology Dispensary, Armavir, Oncologist.
N. Volgin — Main Military Clinical Hospital n.a. Acad. N. N. Burdenko, Moscow, Dermatovenerologist, Doctor of Medical Sciences; E. F. Stranadko — Main Research Center of Laser Medicine, Federal medical-biological Agency of Russia, Moscow, Professor, Doctor of Medical Sciences, R. V. Kagoyants — Armavir Oncology Dispensary, Armavir, Oncologist.
Дата поступления — 9.09.2014 г Дата принятия в печать — 22.09.2014 г.
Волгин В.Н., Странадко Е. Ф., Кагоянц Р.В. Результаты лечения различных морфологических типов рака кожи ЛОР-органов методом фотодинамической терапии. Саратовский научно-медицинский журнал 2014; 10 (3): 555-558.
Цель: оценка эффективности метода фотодинамической терапии (ФДТ) при лечении больных с первичным и рецидивным раком кожи (РК). Материал и методы. В Главном военном клиническом госпитале им. акад. Н. Н. Бурденко лечение РК ЛОР-органов с применением метода ФДТ проведено 108 пациентам. Мужчин было 85 человек, женщин 23. Возраст больных колебался от 43 до 92 лет, составляя в среднем 71 год. Пациентов с базально-клеточным раком кожи (БКРК) ЛОР-органов было 96 человек, пациентов с плоскоклеточным раком кожи (ПКРК) — 8, с метатипическим раком кожи (МТРК) — 4. Показан опыт применения нового перспективного метода ФДТ при лечении больных первичным и рецидивным раком кожи. Результаты. При лечении больных РК ЛОР-органов методом ФДТ полная резорбция (ПР) составила 89,8%, частичная резорбция (ЧР) — 9,3%, без эффекта (БЭ) — 0,9%. Наибольшая эффективность получена при лечении методом ФДТ больных с БКРК ЛОР-органов — 92,7%. Наименее эффективен метод ФДТ при лечении ПКРК ЛОР-органов — 62,5%. При анализе отдаленных результатов рецидивы отсутствовали у 88,7% больных РК ЛОР-органов. В 11,3% случаев возникли рецидивы. Наибольшая эффективность получена при лечении методом ФДТ больных с БКРК ЛОР-органов — 89,9%. Наименее эффективен метод ФДТ при лечении МТРК ЛОР-органов — 66,7%. Заключение.
Мужчин было 85 человек, женщин 23. Возраст больных колебался от 43 до 92 лет, составляя в среднем 71 год. Пациентов с базально-клеточным раком кожи (БКРК) ЛОР-органов было 96 человек, пациентов с плоскоклеточным раком кожи (ПКРК) — 8, с метатипическим раком кожи (МТРК) — 4. Показан опыт применения нового перспективного метода ФДТ при лечении больных первичным и рецидивным раком кожи. Результаты. При лечении больных РК ЛОР-органов методом ФДТ полная резорбция (ПР) составила 89,8%, частичная резорбция (ЧР) — 9,3%, без эффекта (БЭ) — 0,9%. Наибольшая эффективность получена при лечении методом ФДТ больных с БКРК ЛОР-органов — 92,7%. Наименее эффективен метод ФДТ при лечении ПКРК ЛОР-органов — 62,5%. При анализе отдаленных результатов рецидивы отсутствовали у 88,7% больных РК ЛОР-органов. В 11,3% случаев возникли рецидивы. Наибольшая эффективность получена при лечении методом ФДТ больных с БКРК ЛОР-органов — 89,9%. Наименее эффективен метод ФДТ при лечении МТРК ЛОР-органов — 66,7%. Заключение. ФДТ является эффективным методом лечения Рк ЛОР-органов, отличаясь от других методов лечения органосохраняющим характером, отличными косметическими и функциональными результатами, высокой селективностью лечебного действия, отсутствием тяжелого системного влияния на организм. Преимуществом ФДТ по сравнению с другими методами лечения РК ЛОР-органов также является возможность многократного повторения процедуры в случае большого диаметра опухоли и при множественном опухолевом процессе без риска осложнений.
ФДТ является эффективным методом лечения Рк ЛОР-органов, отличаясь от других методов лечения органосохраняющим характером, отличными косметическими и функциональными результатами, высокой селективностью лечебного действия, отсутствием тяжелого системного влияния на организм. Преимуществом ФДТ по сравнению с другими методами лечения РК ЛОР-органов также является возможность многократного повторения процедуры в случае большого диаметра опухоли и при множественном опухолевом процессе без риска осложнений.
Ключевые слова: рак кожи, фотодинамическая терапия, фотосенсибилизаторы.
Целесообразность исследования состава тела с целью оценки и мониторинга нутритивного статуса у детей с дистрофической формой врожденного буллезного эпидермолиза | Макарова
1. Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inheritedepidermolysis bullosa: updated recommendations on diagnosisand classification. J Am Acad Dermatol. 2014;70(6):1103–1126.doi: 10.1016/j.jaad.2014.01.903.
J Am Acad Dermatol. 2014;70(6):1103–1126.doi: 10.1016/j.jaad.2014.01.903.
2. Буллезный эпидермолиз / Под ред. Дж.-Д.Файна, Х. Хинтнера. — М.; 2014. — 357 c.
3. Watkins J. Diagnosis, treatment and management of epidermolysis bullosa. Br J Nurs. 2016;25(8):428–431. doi: 10.12968/ bjon.2016.25.8.428.
4. Макарова С.Г., Намазова-Баранова Л.С., Мурашкин Н.Н., и др. Коррекция нутритивного статуса в комплексной терапии детей, страдающих дистрофической формой врожденного буллезного эпидермолиза диагностика в педиатрии // Педиатрическая фармакология. — 2016. — Т.13. — №6 — С. 577–587. doi: 10.15690/pf.v13i6.1672.
5. Fox AT, Alderdice F, Atherton DJ. Are children with recessive dys-trophic epidermolysis bullosa of low birthweight? Pediatr Dermatol. 2003;20(4):303–306. doi: 10.1046/j.1525-1470.2003.20404.x.
2003;20(4):303–306. doi: 10.1046/j.1525-1470.2003.20404.x.
6. Wells JC, Fewtrell MS. Is body composition important for paediatricians? Arch Dis Child. 2008;93(2):168–172. doi: 10.1136/ adc.2007.115741.
7. Wrottesley SV, Pisa PT, Micklesfield LK, et al. A comparison of body composition estimates using dual-energy X-ray absorptiometry and air-displacement plethysmography in South African neonates. Eur J Clin Nutr. 2016;70(11):1254–1258. doi: 10.1038/ejcn.2016.91.
8. Беляева И.А., Намазова-Баранова Л.С., Тарзян Э.О., и др. Особенности физического развития и состава тканей тела недоношенных детей , получавших различные виды вскармливания (при выписке из стационара 2-го этапа выхаживания) // Вестник Российской академии медицинских наук. — 2014. — Т.69. — №5–6 — С. 71–80. doi: 10.15690/vramn. v69i5-6.1047.
71–80. doi: 10.15690/vramn. v69i5-6.1047.
9. Barbosa-Silva MC, Barros AJ. Bioelectrical impedance analysis in clinical practice: a new perspective on its use beyond body composition equations. Curr Opin Clin Nutr Metab Care. 2005;8(3):311– 317. doi: 10.1097/01.mco.0000165011.69943.39.
10. Böhm A, Heitmann BL. The use of bioelectrical impedance analysis for body composition in epidemiological studies. Eur J Clin Nutr. 2013;67 Suppl 1:S79–85. doi: 10.1038/ejcn.2012.168.
11. Николаев Д.В., Руднев С.Г. Биоимпедансный анализ: основы метода, протокол обследования и интерпретация результатов // Спортивная медицина: наука и практика. — 2012. — №2 — С. 29–36.
12. Николаев Д.В., Смирнов А. В., Бобринская И.Г., Руднев С.Г. Биоимпедансный анализ состава тела человека. — М.: Наука; 2009. — 392 с.
В., Бобринская И.Г., Руднев С.Г. Биоимпедансный анализ состава тела человека. — М.: Наука; 2009. — 392 с.
13. Makarova S, Namazova-Baranova L, Murashkin N, et al. Nutritional status in patients with different forms of epidermolysis bullosa. Arch Dis Child. 2017;102(Suppl 2):A49–A50. doi: 10.1136/ archdischild-2017-313273.127.
14. Fields DA, Gunatilake R, Kalaitzoglou E. Air displacement plethysmography: cradle to grave. Nutr Clin Pract. 2015;30(2):219–226. doi: 10.1177/0884533615572443.
15. Kyle UG, Genton L, Pichard C. Low phase angle determined by bioelectrical impedance analysis is associated with malnutrition and nutritional risk at hospital admission. Clin Nutr. 2013;32(2):294– 299. doi: 10.1016/j.clnu.2012.08.001.
16. Kyle UG, Bosaeus I, De Lorenzo AD, et al. Bioelectrical impedance analysis, part II: utilization in clinical practice. Clin Nutr. 2004;23(6):1430–1453. doi: 10.1016/j.clnu.2004.09.012.
Kyle UG, Bosaeus I, De Lorenzo AD, et al. Bioelectrical impedance analysis, part II: utilization in clinical practice. Clin Nutr. 2004;23(6):1430–1453. doi: 10.1016/j.clnu.2004.09.012.
17. Приказ Минздрава России от 15.11.2012 №920н «Об утверждении Порядка оказания медицинской помощи населению по профилю «диетология» (зарегистрировано в Минюсте России 17.04.2013 №28162).
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом | #01/18
Буллезный эпидермолиз (БЭ) — это группа редких наследственных генетических заболеваний кожи, обусловленных мутациями ряда генов, ответственных за синтез структурных белков кожи. Для заболевания характерна склонность кожи и слизистых оболочек к образованию пузырей, преимущественно на местах незначительного механического воздействия, вследствие нарушения межклеточных связей в эпидермисе или дермоэпидермальном соединении [1, 2]. Наиболее часто пациенты с буллезным эпидермолизом регистрируются в возрасте от 1 до 5 лет. По данным Ю. Ю. Коталевской с соавт., на долю дистрофического варианта приходится больше половины пациентов от общего количества всех форм буллезного эпидермолиза [3]. На сегодняшний день своевременная диагностика и терапия буллезного эпидермолиза остаются одной из главных проблем в медицине. При любой форме буллезный эпидермолиз протекает тяжело, инвалидизация наступает в первые годы течения болезни.
Наиболее часто пациенты с буллезным эпидермолизом регистрируются в возрасте от 1 до 5 лет. По данным Ю. Ю. Коталевской с соавт., на долю дистрофического варианта приходится больше половины пациентов от общего количества всех форм буллезного эпидермолиза [3]. На сегодняшний день своевременная диагностика и терапия буллезного эпидермолиза остаются одной из главных проблем в медицине. При любой форме буллезный эпидермолиз протекает тяжело, инвалидизация наступает в первые годы течения болезни.
Развитие буллезного эпидермолиза обусловлено мутациями генов, кодирующих структурные белки кожи, которые обеспечивают связь между эпидермисом и дермой. К настоящему времени в 15 генах структурных белков кожи выявлено более 1000 мутаций, способных приводить к развитию различных клинических типов врожденного буллезного эпидермолиза [4–6]. С мутациями связаны нарушения синтеза белков: отсутствие белка, синтез функционально неполноценного белка, синтез белка с нарушениями структуры, облегчающими доступ к белку протеаз, что приводит к его быстрому разрушению. Белками, с которыми связано развитие заболевания, являются кератины 5 и 14, десмоплакин, плакофилин-1, плектин, интегрин α6β4, ламинин 332, коллагены VII и XVII типов, киндлин. Эти белки имеют различную локализацию в коже: в кератиноцитах локализуются кератины 5 и 14, внутри светлой пластинки (lamina lucida) базальной мембраны — интегрин α6β4, ламинин 332, коллаген XVII типа, под темной пластинкой (lamina densa) базальной мембраны — коллаген VII типа, на разных уровнях эпидермиса — киндлин [4, 5].
Белками, с которыми связано развитие заболевания, являются кератины 5 и 14, десмоплакин, плакофилин-1, плектин, интегрин α6β4, ламинин 332, коллагены VII и XVII типов, киндлин. Эти белки имеют различную локализацию в коже: в кератиноцитах локализуются кератины 5 и 14, внутри светлой пластинки (lamina lucida) базальной мембраны — интегрин α6β4, ламинин 332, коллаген XVII типа, под темной пластинкой (lamina densa) базальной мембраны — коллаген VII типа, на разных уровнях эпидермиса — киндлин [4, 5].
Выделяют несколько основных типов буллезного эпидермолиза на основе особенностей механизма образования пузыря и клинической картины: простой буллезный эпидермолиз, пограничный буллезный эпидермолиз, дистрофический буллезный эпидермолиз, синдром Киндлера (разный уровень образования пузырей) [7].
Эпидермолиз буллезный простой
Эпидермолиз буллезный простой характеризуется образованием внутриэпидермальных пузырей в результате дезинтеграции и цитолиза кератиноцитов без признаков рубцевания, атрофии и образования милиумов. Тип наследования аутосомно-доминантный. Первые признаки заболевания обычно проявляются на первом году жизни, иногда могут быть уже при рождении ребенка. На месте легкой травматизации, чаще в области кистей, стоп, спины, локтевых и коленных суставов, затылочной области, на неизмененной коже появляются пузыри различных размеров (от 0,5 до 7 см и более) с плотной покрышкой и прозрачным содержимым. Симптом Никольского отрицательный, акантолитические клетки в содержимом пузыря отсутствуют. Через несколько днем пузыри вскрываются, образуя эрозии, покрывающиеся корками и быстро эпителизирующиеся, не оставляя рубцовых изменении кожи или атрофии. Пузырей обычно больше в теплый период года при выраженном гипергидрозе. С возрастом поражения локализуются в основном на конечностях, особенно на стопах и кистях, чему способствует большая травматизация этих участков кожи, тесная, плохо подобранная обувь, а также на участках тесного прилегания одежды. Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается.
Тип наследования аутосомно-доминантный. Первые признаки заболевания обычно проявляются на первом году жизни, иногда могут быть уже при рождении ребенка. На месте легкой травматизации, чаще в области кистей, стоп, спины, локтевых и коленных суставов, затылочной области, на неизмененной коже появляются пузыри различных размеров (от 0,5 до 7 см и более) с плотной покрышкой и прозрачным содержимым. Симптом Никольского отрицательный, акантолитические клетки в содержимом пузыря отсутствуют. Через несколько днем пузыри вскрываются, образуя эрозии, покрывающиеся корками и быстро эпителизирующиеся, не оставляя рубцовых изменении кожи или атрофии. Пузырей обычно больше в теплый период года при выраженном гипергидрозе. С возрастом поражения локализуются в основном на конечностях, особенно на стопах и кистях, чему способствует большая травматизация этих участков кожи, тесная, плохо подобранная обувь, а также на участках тесного прилегания одежды. Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается. Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется. Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера–Коккейна — абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе [8, 12].
Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется. Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера–Коккейна — абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе [8, 12].
Эпидермолиз буллезный соединительный
Эпидермолиз буллезный соединительный характеризуется образованием подэпидермальных пузырей за счет поражения lamina lucida эпидермодермального соединения, расположенной между плазматической мембраной базальных кератиноцитов и базальной мембраной кожи, и развитием атрофических изменений кожи в очагах поражения. Возможна пренатальная диагностика с помощью биопсии кожи 18-недельного плода на основании выявления указанных изменений.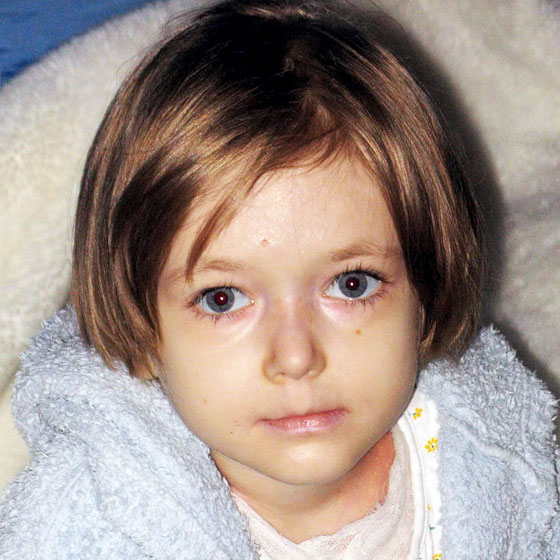 Тип наследования аутосомно-рецессивный.
Тип наследования аутосомно-рецессивный.
Процесс характеризуется появлением пузырей и эрозий уже при рождении ребенка или вскоре после него. В течение нескольких дней процесс генерализуется. Основная локализация высыпаний — кожа груди, головы, слизистые оболочки рта, гортани, трахеи. Хотя кожа кистей и стоп не изменена, ногтевые пластинки дистрофичны, развиваются анонихия, акроостеолиз. Образующиеся на месте пузырей эрозивные поверхности заживают медленно, оставляя участки атрофии кожи. Рубцов и милиумов нет. Многие дети умирают в первые месяцы жизни от сепсиса, анемии [8].
Эпидермолиз буллезный дистрофический
Эпидермолиз буллезный дистрофический характеризуется образованием пузырей вследствие дерматолиза — гибели коллагеновых фибрилл в сосочковом слое дермы ниже lamina densa. Формируются эрозивно-язвенные поверхности, заживающие рубцами, характерны также образование милиумов, изменение ногтей, волос, зубов и другие аномалии.
Эпидермолиз буллезный дистрофический рецессивный генерализованный
Эпидермолиз буллезный дистрофический рецессивный генерализованный (эпидермолиз буллезный дистрофический полидиспластический) отличается образованием пузырей в сосочковом слое дермы и в результате дерматолиза — лизиса коллагеновых фибрилл с фагоцитозом их макрофагами и разрушениями ниже lamina densa. Патологический процесс связывают с увеличением уровня и активности фермента коллагеназы, разрушающей основной компонент опорных коллагеновых фибрилл — коллаген VII (коллагенолиз). Возможна пренатальная диагностика болезни по результатам биопсии кожи плода на 21-й неделе развития и выявления описанных ранее изменений. Первые признаки заболевания появляются уже при рождении (60% больных) или в первые недели жизни. Крупные пузыри, нередко с гемморрагическим содержимым, возникают спонтанно на любом участке кожного покрова и слизистых оболочек. Обширные длительно не заживающие эрозивно-язвенные поверхности, образующиеся при их вскрытии, затрудняют уход и вскармливание новорожденных. Симптом эпидермальной отслойки положительный. На эрозивно-язвенных, нередко кровоточащих, болезненных участках развиваются вегетации. Заживление их происходит медленно, с формированием уродствующих атрофических рубцов. Рубцовые изменения пищевода, глотки, слизистой оболочки рта могут затруднять прием пищи, облитерировать выводные протоки слюнных желез, ограничивать подвижность языка и привести к развитию лейкоплакии.
Патологический процесс связывают с увеличением уровня и активности фермента коллагеназы, разрушающей основной компонент опорных коллагеновых фибрилл — коллаген VII (коллагенолиз). Возможна пренатальная диагностика болезни по результатам биопсии кожи плода на 21-й неделе развития и выявления описанных ранее изменений. Первые признаки заболевания появляются уже при рождении (60% больных) или в первые недели жизни. Крупные пузыри, нередко с гемморрагическим содержимым, возникают спонтанно на любом участке кожного покрова и слизистых оболочек. Обширные длительно не заживающие эрозивно-язвенные поверхности, образующиеся при их вскрытии, затрудняют уход и вскармливание новорожденных. Симптом эпидермальной отслойки положительный. На эрозивно-язвенных, нередко кровоточащих, болезненных участках развиваются вегетации. Заживление их происходит медленно, с формированием уродствующих атрофических рубцов. Рубцовые изменения пищевода, глотки, слизистой оболочки рта могут затруднять прием пищи, облитерировать выводные протоки слюнных желез, ограничивать подвижность языка и привести к развитию лейкоплакии. Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте — от злокачественных новообразований кожи, пищевода, органов полости рта [8].
Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте — от злокачественных новообразований кожи, пищевода, органов полости рта [8].
Эпидермолиз буллезный дистрофический доминантный
Эпидермолиз буллезный дистрофический доминантный (эпидермолиз буллезный дистрофический гиперпластический) характеризуется образованием пузырей в дерме (дерматолиз) ниже lamina densa за счет гибели опорных коллагеновых фибрилл; возможна пренатальная диагностика (по аналогии с дистрофическим полидиспластическим буллезным эпидермолизом).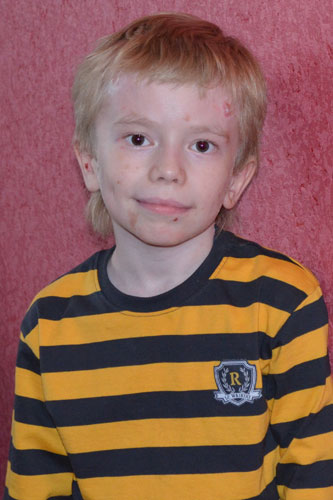 Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4–10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком.
Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4–10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком. У детей более старшего возраста некоторые формы БЭ дифференцируют от доброкачественного буллезного пемфигоида, который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику [8, 9].
У детей более старшего возраста некоторые формы БЭ дифференцируют от доброкачественного буллезного пемфигоида, который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику [8, 9].
На основании только анамнестических и клинических данных не всегда представляется возможным установить диагноз БЭ. У детей старшего возраста в диагностике помогает сбор анамнеза (начало заболевания, значение механического фактора в развитии пузырей и эрозий, постоянно прогрессирующее течение, последовательность развития симптомов, наследственная отягощенность). Обнаружение определенных специфических клинических признаков некоторых форм БЭ может указать на данную патологию, но в большинстве случаев для уточнения диагноза требуется проведение лабораторной диагностики. Традиционное патоморфологическое исследование биоптата кожи не позволяет различить клинические формы БЭ между собой, но при определенном опыте можно отличить по уровню образования пузыря от истинной акантолитической пузырчатки. После исключения всех других возможных заболеваний для подтверждения диагноза заподозренного БЭ и установления его группы могут быть использованы дополнительные методы диагностики: иммунофлюоресцентное антигенное картирование (ИАК), трансмиссионная электронная микроскопия (ТЭМ) и генетический анализ (молекулярная или ДНК-диагностика) [9].
После исключения всех других возможных заболеваний для подтверждения диагноза заподозренного БЭ и установления его группы могут быть использованы дополнительные методы диагностики: иммунофлюоресцентное антигенное картирование (ИАК), трансмиссионная электронная микроскопия (ТЭМ) и генетический анализ (молекулярная или ДНК-диагностика) [9].
Трансмиссионная электронная микроскопия
До начала использования в практике иммунофлюоресцентного анализа ТЭМ являлась «золотым» стандартом в диагностике БЭ, так как позволяла определить уровень образования пузыря и обнаружить ультраструктурные изменения в коже и непосредственно в поврежденных структурах, таких как кератиновые филаменты, десмосомы, полудесмосомы, крепящие (якорные) филаменты, крепящие (якорные) фибриллы, которые при определенных формах БЭ могут отсутствовать, либо содержаться в коже в недостаточном количестве, либо иметь дефектную структуру [9].
Иммунофлюоресцентное антигенное картирование
Иммунофлюоресцентное антигенное картирование было впервые описано в 1981 г. В основе метода лежит последовательная обработка препарата кожи пациента первичными антителами к структурным белкам кожи и вторичными антителами, мечеными флюоресцирующей меткой, которые связываются с первичными антителами. В связи с тем, что известно большое количество белков, дефекты которых приводят к развитию БЭ (кератины 5 и 14, плектин, плакофилин-1, десмоплакин, ламинин 332, интегрин α6β4, коллагены VII и XVII типов, киндлин-1), необходимо определять экспрессию каждого из них. Поэтому для проведения исследования методом ИАК из полученного биоптата кожи изготавливают несколько гистологических препаратов [9–11].
В основе метода лежит последовательная обработка препарата кожи пациента первичными антителами к структурным белкам кожи и вторичными антителами, мечеными флюоресцирующей меткой, которые связываются с первичными антителами. В связи с тем, что известно большое количество белков, дефекты которых приводят к развитию БЭ (кератины 5 и 14, плектин, плакофилин-1, десмоплакин, ламинин 332, интегрин α6β4, коллагены VII и XVII типов, киндлин-1), необходимо определять экспрессию каждого из них. Поэтому для проведения исследования методом ИАК из полученного биоптата кожи изготавливают несколько гистологических препаратов [9–11].
Метод используется для определения уровня образования пузыря (внутриэпидермально, внутри светлой пластинки базальной мембраны, под плотной пластинкой базальной мембраны). ИАК позволяет также определить, дефицит какого из связывающих структуры дермы и эпидермиса белков наблюдается. ИАК определяет экспрессию структурных белков в зоне дермоэпидермального соединения, то есть их присутствие, снижение или отсутствие их экспрессии. Для исследований методом ИАК используют первичные и вторичные моноклональные и поликлональные антитела. Этот метод диагностики более распространен, легче выполним и дешевле по сравнению с ТЭМ. Для правильной постановки диагноза методом ИАК нужно иметь достаточный опыт получения биообразцов кожи, причем одним из требований к получению биообразцов является наличие свежего (не более 24 часов) пузыря на коже. Пузырь может быть как появившимся спонтанно, так и индуцированным намеренно, например, с помощью трения кожи. Биопсию проводят на границе видимо здоровой кожи и свежего пузыря или в зоне трения (через 30 мин после его окончания). Полученный биоптат сразу же подвергается заморозке жидким азотом или помещается в физиологический раствор (0,9% NaCl) на срок не более 24 часов. Для более продолжительного хранения используют специальную транспортную среду Michel. В таком состоянии биообразцы кожи могут сохраняться в течение нескольких недель [12].
Для исследований методом ИАК используют первичные и вторичные моноклональные и поликлональные антитела. Этот метод диагностики более распространен, легче выполним и дешевле по сравнению с ТЭМ. Для правильной постановки диагноза методом ИАК нужно иметь достаточный опыт получения биообразцов кожи, причем одним из требований к получению биообразцов является наличие свежего (не более 24 часов) пузыря на коже. Пузырь может быть как появившимся спонтанно, так и индуцированным намеренно, например, с помощью трения кожи. Биопсию проводят на границе видимо здоровой кожи и свежего пузыря или в зоне трения (через 30 мин после его окончания). Полученный биоптат сразу же подвергается заморозке жидким азотом или помещается в физиологический раствор (0,9% NaCl) на срок не более 24 часов. Для более продолжительного хранения используют специальную транспортную среду Michel. В таком состоянии биообразцы кожи могут сохраняться в течение нескольких недель [12].
Генетический анализ (молекулярная или ДНК-диагностика)
Генетический анализ (молекулярная или ДНК-диагностика) является оптимальным методом для определения типа наследования и специфических мутаций, имеющихся у больных БЭ, а также наиболее точным методом для верификации различных клинических форм простого, пограничного и дистрофического БЭ. Поиск известных мутаций является первичным диагностическим подходом, особенно в семьях больных БЭ с аутосомно-доминантным типом наследования, в силу того, что данные мутации, как правило, идентичны в семье, страдающей данным заболеванием. Также имеются данные, что поиск неизвестных мутаций может быть осуществлен с использованием ДНК-амплификационного метода и секвенированием РНК дефектных генов [9].
Поиск известных мутаций является первичным диагностическим подходом, особенно в семьях больных БЭ с аутосомно-доминантным типом наследования, в силу того, что данные мутации, как правило, идентичны в семье, страдающей данным заболеванием. Также имеются данные, что поиск неизвестных мутаций может быть осуществлен с использованием ДНК-амплификационного метода и секвенированием РНК дефектных генов [9].
Пренатальная диагностика
Как известно, до настоящего времени не разработано эффективных методов терапии БЭ, поэтому особенно важное значение на стадии планирования семьи, находящейся в группе риска наследования данной патологии, приобретает пренатальная диагностика и генетическое консультирование. Первое сообщение о пренатальной диагностике одной из наиболее тяжелых форм БЭ (летального генерализованного БЭ Герлитца) было опубликовано еще в 1980 г. [13].
Усовершенствование молекулярных методов диагностики позволило проводить пренатальную диагностику на основании анализа фетальной ДНК, которую можно получить из амниотической жидкости и/или ворсинок хориона. Биопсия ворсин хориона и амниоцентез позволяют существенно снизить риск преждевременного прерывания беременности по сравнению с фетоскопией, а также провести данный вид диагностики в I триместре (до 11 недель беременности). Дородовая диагностика возможна только в том случае, когда ее планируют заранее. До наступления беременности нужно установить точно генетический дефект у болеющего члена семьи. Если дефектный ген уже известен, то поиск такой же поломки у плода занимает всего несколько дней, что создает возможность проведения аборта в безопасные для беременной женщины сроки. Трудностью в диагностике может являться мозаицизм, при котором у ребенка могут присутствовать две и более генетически различные популяции клеток [14, 15].
Биопсия ворсин хориона и амниоцентез позволяют существенно снизить риск преждевременного прерывания беременности по сравнению с фетоскопией, а также провести данный вид диагностики в I триместре (до 11 недель беременности). Дородовая диагностика возможна только в том случае, когда ее планируют заранее. До наступления беременности нужно установить точно генетический дефект у болеющего члена семьи. Если дефектный ген уже известен, то поиск такой же поломки у плода занимает всего несколько дней, что создает возможность проведения аборта в безопасные для беременной женщины сроки. Трудностью в диагностике может являться мозаицизм, при котором у ребенка могут присутствовать две и более генетически различные популяции клеток [14, 15].
Преимплантационная генетическая диагностика
Преимплантационная генетическая диагностика является перспективным альтернативным методом наряду с вышеперечисленными, кроме того, метод может быть частью экстракорпорального оплодотворения, выявляя тем самым различные отклонения развития эмбриона еще на стадии бластоцисты. Суть данного метода заключается в оплодотворении овоцита (незрелой яйцеклетки) in vitro, что ведет к образованию эмбриона. Когда эмбрион достигает стадии 8- или 12-клеточного субъекта, производится отщепление одной клетки для дальнейшего генетического анализа. Если генетический анализ указывает на нормальный генотип — эмбрион подсаживают в матку, иначе его утилизируют [16].
Суть данного метода заключается в оплодотворении овоцита (незрелой яйцеклетки) in vitro, что ведет к образованию эмбриона. Когда эмбрион достигает стадии 8- или 12-клеточного субъекта, производится отщепление одной клетки для дальнейшего генетического анализа. Если генетический анализ указывает на нормальный генотип — эмбрион подсаживают в матку, иначе его утилизируют [16].
Неинвазивные методы пренатальной диагностики
Все вышеперечисленные методы пренатальной диагностики являются инвазивными и в той или иной степени могут повлиять на развитие плода и на течение беременности. В связи с этим идет постоянный поиск новых методов, снижающих риск инвазивного воздействия [9].
Среди таких методов можно выделить метод ультразвуковой диагностики, который позволяет визуализировать некоторые особенности развития плода при ограниченном количестве тяжелых патологий, а также другие структуры содержимого матки, которые могут быть идентификаторами некоторых врожденных заболеваний. К таким показателям можно отнести особенности фенотипа плодного яйца: размеры носовой кости, воротниковой области, желточного мешка [9].
Генетическое консультирование лучше проводить дерматологом или генетиком, специализирующимся на БЭ. В конечном итоге диагноз ставится на основании клинического фенотипа, способа наследования, а также, если возможно, провести мутационный анализ пробанда.
В периоде новорожденности наблюдение и симптоматическая терапия за больными врожденным буллезным эпидермолизом (ВБЭ) проводится в условиях отделения интенсивной терапии педиатрического стационара. Основные принципы терапии не отличаются от таковых у взрослых лиц. Лекарственная терапия проводится с учетом возрастных ограничений к назначению лекарственных препаратов. Профилактические прививки противопоказаны только в период нарушения общего состояния ребенка. Беременность у больных ВБЭ обычно протекает без осложнений. При планировании беременности необходимо провести лечение сопутствующих заболеваний, в том числе очагов хронической инфекции, хирургическое устранение контрактур и псевдосиндактилии. Во избежание травмирования принимают меры предосторожности при взятии крови, вагинальном исследовании, пальпации, УЗИ. Основным в период беременности является наружное лечение [7].
Больным буллезным эпидермолизом необходимо избегать физических нагрузок, связанных с повышением потоотделения, травмоопасных ситуаций, резких движений. Перевязочные материалы, одежда, закрытая обувь позволяют свести к минимуму травмирование кожи. При хорошем самочувствии и отсутствии высыпаний на коже допустимо плавание.
Тяжелые подтипы БЭ, а также нетяжелые, но протекающие с поражением полости рта, требуют особого внимания к питанию. Диета должна быть механически, термически и химически щадящей (протертой и полужидкой, не горячей). Питание особенно важно для больных с большой площадью поражения кожи, так как они теряют питательные вещества и влагу с тканевой жидкостью, необходимой для заживления ран и борьбы с инфицированием. Питание больных должно быть богато белками, углеводами, жирами, а также содержать витамины, минералы, пищевые волокна и большое количество жидкости. При однообразной диете и недостаточном питании потребление витаминов и минералов с пищей ограничено, поэтому рекомендуется дополнительно принимать поливитаминно-минеральные комплексы. При наличии эрозий во рту, дисфагии и сужении пищевода назначают жидкие комплексы. В их составе особенно важны витамины А, группа В, С, D и Е, из минералов — железо, цинк, селен и кальций: витамин А (ретинола пальмитат), масляный раствор 100 000 МЕ/мл перорально на ночь по 10–30 капель (в зависимости от возраста и веса пациента) в течение 2 месяцев. Курсы терапии можно повторять с интервалом в 3 месяца; витамин С (аскорбиновая кислота) 100 мг перорально после еды 3–4 раза в сутки в течение 2 недель. При большом количестве пузырей и эрозий пациент нуждается в восполнении теряемой жидкости [7, 17].
В настоящее время пренатальная диагностика является лучшим способ профилактики тяжелых наследственных заболеваний. Она возможна только при планировании беременности задолго до ее наступления. Медико-генетическое консультирование семьи проводится с целью оценки риска появления больного ребенка в семье, информирования семьи о риске развития наследственного заболевания, о возможных диагностических и терапевтических методах. Показания для консультирования — предыдущее рождение ребенка с ВБЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
У пациента с БЭ профилактические меры в отношении появления пузырей на коже и слизистых оболочках включают ограничение возможности травмирования кожи (одежда, диета, особенности ухода, неадгезивные повязки, наружные средства, уход за полостью рта). Таким образом, профилактика развития осложнений, диспансеризация, периодический контроль лабораторных показателей для выявления и контроля анемии, полный осмотр пациентов с целью раннего выявления злокачественных опухолей кожи, своевременное лечение зубов будут способствовать существенному облегчению и улучшению качаства жизни пациентов.
Литература
- Lara-Corrales I., Mellerio J. E., Martinez A. E. et al. Dilated cardiomyopathy in epidermolysis bullosa: a retrospective, multicenter study // Pediatr Dermatol. 2010; 27 (3): p. 238–243.
- Гараева З. Ш., Юсупова Л. А., Мавлютова Г. И., Юнусова Е. И., Мирзина Д. Р. Буллезный эпидермолиз. Сборник материалов Всероссийской научно-практической конференции с международным участием «Казанские дерматологические чтения: синтез науки и практики». Казань, 2016. С. 8–20.
- Коталевская Ю. Ю., Кропачева В. В., Марычева Н. М. Буллезный эпидермолиз — состояние проблемы в России. Материалы I Евразийской Конференции по редким заболеваниям и редким лекарствам и III Всероссийской Конференции по редким заболеваниям и редко применяемым медицинским технологиям «Дорога жизни». М., 2012. С. 15–19.
- Mitsuhashi Y., Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa // Arch Dermatol Res. 2003; 295 (Suppl 1.): 29–33.
- Uitto J., Richard G. Progress in epidermolysis bullosa: genetic classification and clinical implications // Am J Med Genet C Semin Med Genet. 2004; 131 C (1): 61–74.
- Юсупoвa Л. A. Иммунопатология хронических дерматозов. Казань: НБ КГМА, 2017. 108 с.
- Федеральные клинические рекомендации. Дерматовенерология 2015: Болезни кожи. Инфекции, передаваемые половым путем. 5-е изд., перераб. и доп. М.: Деловой экспресс. 2016. 768 с.
- Иванов О. И. Кожные и венерические болезни: Шико. М., 2006. 133–137 с.
- Альбанова В. И., Чикин В. В., Епишев Р. В. К вопросу о диагностике врожденного буллезного эпидермолиза // Вестник дерматологии и венерологии. 2014. № 3. С. 53–59.
- Pohla-Gubo G., Cepeda-Valdes R., Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 201–210.
- Cepeda-Valdés R., Pohla-Gubo G., Borbolla-Escoboza J. R. et al. Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa // Actas Dermosifiliogr. 2010; 101 (8): p. 673–682.
- Intong L. R., Murrell D. F. How to take skin biopsies for epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 197–200.
- Rodeck C. H., Eady R. A., Gosden C. M. Prenatal diagnosis of epidermolysis bullosa letalis // Lancet. 1980; 1: p. 949–952.
- Pasmooij A. M., Pas H. H., Bolling M. C., Jonkman M. F. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3 // J Clin Invest. 2007; 117 (5): p. 1240–1248.
- Pasmooij A. M., Pas H. H., Deviaene F. C. et al. Multiple correcting COL17 A1 mutations in patients with revertant mosaicism of epidermolysis bullosa // Am J Hum Genet. 2005; 77 (5): p. 727–740.
- Renwick P., Ogilvie C. M. Preimplantation genetic diagnosis for monogenic diseases: overview and emerging issues // Expert Rev Mol Diagn. 2007; 7: p. 33–43.
- Boeira V. L., Souza E. S., Rocha Bde O. et al. Inherited epidermolysis bullosa: clinical and therapeutic aspects // An Bras Dermatol 2013; 88 (2): p. 185–198.
Л. А. Юсупова*, 1, доктор медицинских наук, профессор
Е. И. Юнусова*, кандидат медицинских наук
З. Ш. Гараева*, кандидат медицинских наук
Г. И. Мавлютова*, кандидат медицинских наук
М. А. Морозова**
* ГБОУ ДПО КГМА МЗ РФ, Казань
** ГАУЗ РККВД, Казань
1 Контактная информация: [email protected]
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом/ Л. А. Юсупова, Е. И. Юнусова, З. Ш. Гараева, Г. И. Мавлютова, М. А. Морозова
Для цитирования: Лечащий врач № 1/2018; Номера страниц в выпуске: 71-74
Теги: заболевания кожи, генетические мутации, белки
ПЛОСКОКЛЕТОЧНЫЙ РАК КОЖИ У БОЛЬНЫХ РЕЦЕССИВНЫМ ДИСТРОФИЧЕСКИМ БУЛЛЕЗНЫМ ЭПИДЕРМОЛИЗОМ: СЛУЧАИ С АГРЕССИВНЫМ ТЕЧЕНИЕМ НОВООБРАЗОВАНИЯ
Fine J.D., Bruckner-Tuderman L., Eady R.A. et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70 (6): 1103-1126. doi: 10.1016/j.jaad.2014.01.903.
Chung H.J., Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin. 2010; 28 (1): 93-105. doi: 10.1016/j. det.2009.10.011
Watanabe M., Natsuga K., Shinkuma S., Shimizu H. Epidermal aspects of type VII collagen: implications for dystrophic epidermolysis bullosa and epidermolysis bullosa acquisita. J Dermatol. 2018; 45 (5): 515-521. doi: 10.1111/1346-8138.14222.
Кубанов А.А., Карамова А.Э., Альбанова В.И. и др. Врожденный буллезный эпидермолиз: особенности регенерации эпидермиса и методы терапии. Вестник дерматологии и венерологии 2017; (4): 28-37.
Eming S.A., Martin P., Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 2014, 6 (265): 265sr6. doi: 10.1126/scitrans-lmed.3009337.
Cianfarani F., Zambruno G., Castiglia D., Odorisio T. Pathomechanisms of altered wound healing in recessive dystrophic epidermolysis bullosa. Am. J. Pathol. 2017; 187 (7): 1445-1453. doi: 10.1016/j.ajpath.2017.03.003.
Nystram A., Bruckner-Tuderman L. Injury- and inflammation-driven skin fibrosis: the paradigm of epidermolysis bullosa. Matrix Biol. 2018; 68-69: 547-560. doi: 10.1016/j.matbio.2018.01.016.
Guerra L., Odorisio T, Zambruno G., Castiglia D. Stromal microenvironment in type VII collagen-deficient skin: the ground for squamous cell carcinoma development. Matrix Biol. 2017; 63: 1-10. doi: 10.1016/j.matbio.2017.01.002.
Kawasaki H., Sawamura D., Iwao F. et al. Squamous cell carcinoma developing in a 12-year old boy with non-Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2003; 148 (5): 1047-1050. doi: 10.1046/j.1365-2133.2003.05270.x.
Fine J.D., Johnson L.B., Weiner M. et al. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986-2006. J. Am. Acad. Dermatol. 2009; 60 (2): 203-211. doi: 10.1016/j. jaad.2008.09.035.
Venables Z.C., Nijsten T., Wong K.F. et al. Epidemiology of basal and cutaneous squamous cell carcinoma in the U.K. 2013-15: A cohort study. Br. J. Dermatol. 2019; 181 (3): 474-482. doi: 10.1111/bjd.17873.
Que S.K., Zwald F.O., Schmults C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018; 78 (2): 237247. doi: 10.1016/j.jaad.2017.08.059.
Stang A., Khil L., Kajuter H. et al. Incidence and mortality for cutaneous squamous cell carcinoma: comparison across three continents. J Eur Acad Dermatol Venereol. 2019; 33 Suppl 8: 6-10. doi: 10.1111/jdv.15967.
Беляев А.М., Прохоров Г.Г., Раджабова З.А., Байкалова О.И. Обзор современных методов лечения плоскоклеточного рака кожи. Вопросы онкологии. 2019; 65 (1): 7-15.
Condorelli A.G., Dellambra E., Logli E. et al. Epidermolysis bullosa-associated squamous cell carcinoma: from pathogenesis to therapeutic perspectives. Int J Mol Sci. 2019; 20 (22): 5707. doi: 10.3390/ijms20225707.
Mellerio J.E., Robertson S.J., Bernardis C. et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: Best clinical practice guidelines. Br. J. Dermatol. 2016; 174 (1): 56-67. doi: 10.1111/ bjd.14104.
Карамова А.Э., Чикин В.В., Альбанова В.И. и др. Плоскоклеточный рак кожи, развившийся у больной рецессивным дистрофическим буллезным эпидермолизом. Вестник дерматологии и венерологии. 2016; (3): 83-89.
Кубанов А.А., Карамова А.Э., Чикин В.В., и др. Эпидемиология и состояние оказания медицинской помощи больным врожденным буллезным эпидермолизом в Российской Федерации. Вестник Российской академии медицинских наук. 2018; 73 (6): 420-430.
19. Kelly-Mancuso G., Kopelan B., Azizkhan R.G., Lucky A.W. Junctional Epidermolysis Bullosa Incidence and Survival: 5-Year Experience of the Dystrophic Epidermolysis Bullosa Research Association of America (DebRA) Nurse Educator, 2007 to 2011. Pediatr Dermatol 2013; 31 (2): 159-162. doi: 10.1111/pde.12157.
Врожденный буллезный эпидермолиз: вертикальный перенос материнских аутоантител от матери к младенцу | Врожденные пороки | JAMA дерматология
Приобретенный буллезный эпидермолиз (EBA) — это редкий хронический аутоиммунный буллезный дерматоз, который вызывается аутоантителами против неколлагенового конца α-цепи коллагена типа VII, что приводит к уменьшению заякоривающих фибрилл в lamina densa. Насколько нам известно, случай, связанный с новорожденным с врожденной EBA, еще не описан в литературе.Мы описываем новорожденного с преходящей EBA из-за пассивной передачи материнских аутоантител.
32-летняя женщина с беременностью 4, параграф 3 родила девочку, у которой при рождении были обнаружены напряженные волдыри и участки обнаженной кожи. Анамнез родов отличался индукцией родов на 36 неделе беременности из-за маловодия и ограничения внутриутробного развития. Новорожденный при родах был энергичным и активным, вмешательства не потребовалось.Оценка по шкале Апгар составила 9 баллов на 1 и 5 минуте. На 1-й день жизни отмечалось прогрессирование кожных поражений пациента с развитием новых булл и плохим питанием. Она была переведена в отделение интенсивной терапии новорожденных, проконсультирована со службой дерматологии.
При физикальном обследовании пациентка выглядела хорошо, хотя и была маленькой для гестационного возраста. У нее были множественные поверхностные и глубокие эрозии на лице, груди, животе и конечностях (рис. 1A), наиболее заметные поражения были на руках, лодыжках и ступнях (рис. 1B).У нее также были разбросаны неповрежденные пузырьки и пузыри. На ее губах и правом ярусе были обнаружены пузырьки, эрозии и легкая корка. Слизистая оболочка рта была чистой, на ногтях было небольшое подногтевое кровоизлияние.
Рисунок 1
Фотографии, изображающие первоначальное клиническое проявление нашего пациента с эрозиями и волдырями и последующее разрешение в возрасте 2 месяцев. А. Множественные поверхностные и глубокие эрозии и неповрежденные пузыри на груди, животе и конечностях.B. Большие глубокие эрозии и обнажение лодыжек и стоп. В. Зажившие эрозии с гипопигментацией, рубцами и милиумами на лодыжках и ступнях.
Мать больной поставлена диагнозом ВБА. Во время родов у матери были множественные поверхностные эрозии кожи с легкими корками, гипопигментацией, гиперпигментацией и рубцами на двусторонней передней поверхности голеней, тыльной стороне рук и ступнях. Присутствовала ониходистрофия, целых булл не было.
Предыдущая диагностическая оценка матери, которая была завершена годом ранее в другом учреждении, включала биопсию кожи, иммунофлуоресцентные исследования и иммуноферментный анализ (ELISA). Гистопатологическая оценка показала фиброзную грануляционную ткань с вышележащим умеренно реактивным эпидермисом. Прямая иммунофлуоресценция выявила отложение IgG, IgM и C3 в дермоэпидермальном соединении без отложения иммуноглобулинов в эпидермальном межклеточном пространстве.Результаты анализов ELISA на уровни антител IgG BP180 и IgG BP230 были нормальными. Уровень антинуклеарных антител был повышен лишь незначительно, 1: 160, с однородной картиной. Эти клинические, гистопатологические и лабораторные данные убедительно свидетельствуют в пользу диагноза EBA.
У матери пациентки в анамнезе были стриктуры пищевода и поражение глаз, и в прошлом она лечилась иммунодепрессантами, включая циклофосфамид и периодическую пероральную терапию преднизоном, при обострениях ее заболевания.Она прекратила иммуносупрессивную терапию в первом триместре беременности. Трое других детей, родившихся до того, как ей поставили диагноз EBA, были здоровы и не имели проблем с пузырями. Другой семейной истории буллезного расстройства не было. Результаты обследования матери на предмет сопутствующих состояний были нормальными.
Учитывая клиническую картину нашей пациентки и диагноз ее матери, мы диагностировали вероятную неонатальную ЭБА в результате пассивного переноса материнских антител.Для подтверждения диагноза были выполнены биопсия кожи, иммунофлуоресцентные исследования, ELISA и иммуноблот-анализ. Гистопатологическое исследование свежей волдыря на коже левого бедра показало слабовоспалительный субэпидермальный волдырь (рис. 2А). Прямая иммунофлуоресценция перилезионной кожи выявила линейное отложение IgG1, IgG4, IgM и C3 на дермальной стороне дермоэпидермального соединения (рис. 2В). Окраски были отрицательными на IgA и фибриноген, а в межклеточном эпидермальном пространстве не было отложений.
Рисунок 2
Результаты гистопатологического исследования и иммунофлуоресцентного исследования младенца соответствовали буллезному эпидермолизу. A, Многоклеточный субэпидермальный волдырь (гематоксилин-эозин, исходное увеличение × 20). B: прямая иммунофлуоресценция, показывающая линейное отложение IgG1, IgG4, IgM и C3 в дермоэпидермальном соединении на дермальной стороне волдыря (гематоксилин-эозин, исходное увеличение × 40). C. Непрямая иммунофлуоресценция, показывающая положительное окрашивание с IgG на дермоэпидермальном соединении на дермальной стороне волдыря при разведении 1:40 (гематоксилин-эозин, исходное увеличение × 20).
Постепенное разведение образца сыворотки младенца от 1:10 до 1: 5000, нанесенного на расщепленную солью кожу приматов и окрашенного флуоресцеин-конъюгированными козьими антителами против IgG человека, показало положительное окрашивание IgG в дермоэпидермальном соединении с участием дермальной стороны для всех разведения от 1:10 до 1:40 (Рисунок 2C). Анализ ELISA (фиг. 3) был выполнен с использованием С-концевого неколлагенового (NC1) домена коллагена типа VII в качестве субстрата 1 и разведения образца сыворотки пациента 1: 100.Связанные аутоантитела выявляли с помощью конъюгированных с щелочной фосфатазой козьих антител против человеческого IgG. Два нормальных образца человека (NHS1 и NHS2), которые использовались в качестве контроля, показали очень низкую реактивность со значениями менее 0,18 OD (оптическая плотность). В отличие от контрольных образцов, образец сыворотки нашего пациента содержал антитела к коллагену VII типа на уровнях выше, чем в образце сыворотки пациента с известным EBA (положительный контроль). Чтобы дополнительно подтвердить результаты ELISA, мы провели иммуноблот-анализ с использованием рекомбинантного NC1.Как показано на фиг. 4, рекомбинантный белок NC1 массой 145 кДа распознавался образцом сыворотки нашего пациента и образцом положительного контроля, но не 2 образцами отрицательного контроля при разведении 1:50.
Рисунок 3
Иммуноферментный анализ. На оси ординат OD (оптическая плотность) коррелирует с присутствием антител к коллагену VII типа; OD измеряли по оптической плотности при длине волны 405 нм. Два нормальных образца сыворотки человека (NHS1 и NHS2) показали очень низкую реактивность.Образец сыворотки нашего пациента содержал антитела к коллагену VII типа на уровнях выше, чем в образце сыворотки пациента с приобретенным буллезным эпидермолизом (EBA) (положительный контроль).
Рисунок 4
Иммуноблот-анализ с использованием рекомбинантного С-концевого неколлагенового домена (NC1) коллагена типа VII. Пациент указывает на новорожденного, описанного в этой статье. В качестве положительного контроля использовали известного пациента с приобретенным буллезным эпидермолизом (EBA), а в качестве отрицательного контроля использовали 2 нормальных образца сыворотки крови человека (NHS1 и NHS2).
Учитывая ожидаемое спонтанное снижение уровня циркулирующих антител у нашей пациентки, мы отказались от иммуносупрессивной терапии и выбрали поддерживающую терапию с тщательным мониторингом ее боли и статуса питания. Она очень хорошо справилась с тщательным уходом за ранами, мазью с бацитрацином для открытых участков, пропитанными петролатумом повязками и марлевыми повязками. Ее первоначальное кормление было плохим, но, учитывая возможность повреждения слизистой оболочки, мы не рекомендовали установку зонда через желудочный зонд.Ей внутривенно вводили жидкости и разрешали кормить без ограничений стандартной смесью и обычной детской соской. Ее боль хорошо контролировалась с помощью режима ацетаминофена и пероральной сахарозы перед сменой повязки; ей потребовался пероральный морфин только один раз. Во время недельной госпитализации новые волдыри появлялись редко, и ее питание значительно улучшилось.
В возрасте 3 недель ее губы, слизистые оболочки полости рта и носа выглядели нормальными. На тыльной стороне рук были обнаружены хорошо заживающие эрозии и дисхромия с легким подногтевым кровоизлиянием на миниатюрах.Были стойкие поверхностные эрозии с шелушением на лодыжках и стопах, но новых волдырей не было. В возрасте 2 месяцев все эрозии полностью зажили, остались только рубцы, гиперпигментация и гипопигментация. Области предыдущих булл теперь были усеяны множественными милиумами (рис. 1С).
Приобретенный буллезный эпидермолиз — это аутоиммунное буллезное заболевание, которое вызывается циркулирующими аутоантителами IgG, направленными против домена 145 кДа NC1 коллагена VII. 2 , 3 Woodley et al. 4 предоставили прямые доказательства того, что человеческие аутоантитела являются патогенными для EBA, когда безволосым иммунокомпетентным взрослым мышам, которым вводили аффинно очищенные антитела к коллагену VII типа из сыворотки пациентов с EBA, развились клинические, гистологические исследования. , иммунологические и ультраструктурные параметры, соответствующие EBA. IgG1 и IgG4 являются основными подклассами антител в EBA. 5 , 6 Период полувыведения IgG из плазмы составляет приблизительно 14 дней у среднего взрослого человека.15-дневный период полувыведения IgG к BP180 был зарегистрирован у новорожденного с гестационным пемфигоидом. 7 Высыпания у этого новорожденного исчезли в возрасте 8 дней, раньше, чем ожидалось исчезновение активности против ВР180. У нашего пациента не появилось новых волдырей примерно через 10 дней жизни.
Клинически существует 2 фенотипа EBA. Воспалительный тип имитирует другие буллезные заболевания, включая буллезный пемфигоид, буллезную болезнь с линейным IgA и рубцовый пемфигоид.EBA классического типа является более распространенным и фенотипически сходным с дистрофическим буллезным эпидермолизом, с хрупкостью кожи, образованием пузырей, рубцами и милиумами, вызванными травмой. 8 Слизистые оболочки ротовой полости и глаз могут поражаться по-разному. Приобретенный буллезный эпидермолиз встречается редко, с оценочной заболеваемостью 0,25 на миллион в Западной Европе, 8 , и большинство случаев приобретается во взрослом возрасте.
Сообщается о
EBA «Детство». 8 -12 Обычно дети младше 16 лет.В отличие от болезни взрослых, детские болезни чаще являются воспалительными, 8 имеет повышенную частоту поражения слизистых оболочек, 13 , 14 показывает лучший ответ на лечение и, следовательно, имеет лучший долгосрочный прогноз. 15 , 16 Лечение детей состоит из преднизона или дапсона, отдельно или в комбинации. Последующее наблюдение ограничено редкостью заболевания; тем не менее, долгосрочный прогноз для детской EBA кажется благоприятным. Положительный ответ на лечение и ремиссия заболевания наблюдаются в течение нескольких месяцев после начала системной терапии. 15
Преходящие аутоиммунные буллезные кожные заболевания новорожденных встречаются редко. Предполагаемый механизм заболевания — пассивный перенос аутоантител материнского IgG через ткани плаценты. Новорожденные, рожденные с преходящими пузырями, были зарегистрированы у матерей с вульгарной пузырчаткой 17 , 18 (включая тех, у которых на момент родов не было клинических симптомов 19 ), листовидной пузырчаткой, 20 , 21 и гестационным пемфигоидом. . 22 Аутоиммунные буллезные заболевания могут модулироваться беременностью, предположительно вторичной по отношению к гормональным воздействиям эстрогена и прогестерона, с проявлением болезни во время беременности или сразу после родов. 23 Было показано, что количество патогенных антител к ВР180 увеличивается непосредственно до и после родов у пациентов с гестационным пемфигоидом, предположительно в ответ на высвобождение антигена ВР180 из хориональной мембраны. 24 Точная роль гормонов в ускорении образования пузырей или титре патогенных антител неизвестна.
Насколько нам известно, подобных исследований у беременных женщин с ЭБА не проводилось. В литературе описаны две пациентки, в том числе 1 пациентка, у которой развилась ВБА на 2-й день после родов, с исчезновением пузырей во время менопаузы, 25 и 1 пациентка, у которой был рецидив ВБА в течение первого месяца гестации, с выраженными симптомами. улучшение ее кожи после прерывания беременности. 26
Насколько нам известно, ранее не сообщалось о врожденной EBA, вероятно, потому, что сама EBA является чрезвычайно редким заболеванием.Опыт с пузырчаткой новорожденных показал, что такое вертикально передающееся аутоиммунное образование пузырей, по-видимому, самоограничивается и разрешается при поддерживающей терапии. 18 После 10-дневного возраста у нашего пациента не было новых волдырей, и все эрозии полностью зажили к 2-месячному возрасту. Поскольку это первый зарегистрированный случай неонатальной ЭБА (насколько нам известно), делать выводы относительно долгосрочного прогноза преждевременно.
Для корреспонденции: Энтони Дж.Манчини, доктор медицины, отделение детской дерматологии, Детская мемориальная больница, 2300 Children’s Plaza, Ste 107, Chicago, IL 60614 ([email protected]).
Принята к публикации: 31 августа 2010 г.
Опубликовано в сети: 15 ноября 2010 г. doi: 10.1001 / archdermatol.2010.317
Вклад авторов: Доктора Абрамс, Смидт, Бенджамин и Манчини имели полный доступ ко всем данным в исследовании и несли ответственность за целостность данных и точность анализа данных. Концепция и дизайн исследования : Абрамс, Шмидт, Бенджамин и Манчини. Сбор данных : Абрамс, Смидт, Бенджамин, Чен, Вудли и Манчини. Анализ и интерпретация данных : Абрамс, Смидт, Бенджамин, Чен, Вудли и Манчини. Составление рукописи : Абрамс и Манчини. Критический пересмотр рукописи для важного интеллектуального содержания : Абрамс, Шмидт, Бенджамин, Чен, Вудли и Манчини. Административная, техническая и материальная поддержка : Абрамс и Манчини. Наблюдение за учебой : Манчини.
Раскрытие финансовой информации: Не сообщалось.
Дополнительные вклады: Марьян Мирзабейги, доктор медицины, предоставил дерматопатологические слайды и фотографии.
1. Чэнь
MChan
LSCai
XO’Toole
EASample
Дж. К. Вудли
DT Разработка ELISA для быстрого обнаружения аутоантител к коллагену VII типа при приобретенном буллезном эпидермолизе. J Invest Dermatol 1997; 108
(1)
68-72PubMedGoogle ScholarCrossref 2. Вудли
DTBriggaman
РАО’Киф
EJInman
AOQueen
LLGammon
WR Идентификация аутоантигена базальной мембраны кожи при приобретенном буллезном эпидермолизе. N Engl J Med 1984; 310
(16)
1007-1013PubMedGoogle ScholarCrossref 3. Вудли
DTBurgeson
RELunstrum
GBruckner-Tuderman
LReese
MJBriggaman
Антиген буллезного эпидермолиза RA является глобулярным карбоксильным концом проколлагена VII типа. Дж. Клин Инвест 1988; 81
(3)
683-687PubMedGoogle ScholarCrossref 4. Вудли
DTRam
RDoostan
А
и другие. Индукция приобретенного буллезного эпидермолиза у мышей путем пассивной передачи аутоантител от пациентов. J Invest Dermatol 2006; 126
(6)
1323–1330PubMedGoogle ScholarCrossref 5. Эдвардс
SWakelin
SHWojnarowska
F
и другие. Буллезный пемфигоид и приобретенный буллезный эпидермолиз: клинические проявления, прогноз и иммунопатология у 11 детей. Педиатр дерматол 1998; 15
(3)
184-190PubMedGoogle ScholarCrossref 6. Бернар
PProst
Каукутюрье
PDurepaire
NDenis
FBonnetblanc
JM Распределение подклассов аутоантител IgG при рубцовом пемфигоиде и приобретенном буллезном эпидермолизе. J Invest Dermatol 1991; 97
(2)
259-263PubMedGoogle ScholarCrossref 7. Аояма
Ясаи
Хиоки
К.Фунато
MKondo
NKitajima
Y Герпес у матери и новорожденного: иммуноклиническая перспектива, основанная на еженедельном наблюдении за индексом неколлагенового домена буллезного пемфигоидного антигена в иммуноферментном анализе. Arch Dermatol 2007; 143
(9)
1168–1172PubMedGoogle ScholarCrossref 8.Маюзуми
Макияма
МНишие
W
и другие. Приобретенный буллезный эпидермолиз у детей с аутоантителами против неколлагеновых 1 и 2 доменов коллагена типа VII: отчет о клиническом случае и обзор литературы. Br J Дерматол 2006; 155
(5)
1048-1052PubMedGoogle ScholarCrossref 9.Bernard
PVaillant
LLabeille
B
и другие. Французская исследовательская группа по буллезным заболеваниям, Заболеваемость и распространение субэпидермальных аутоиммунных буллезных кожных заболеваний в трех регионах Франции. Arch Dermatol 1995; 131
(1)
48-52PubMedGoogle ScholarCrossref 10. Калло-Мелло
CBodemer
CCaux
F
и другие. Приобретенный буллезный эпидермолиз в детском возрасте. Arch Dermatol 1997; 133
(9)
1122–1126PubMedGoogle ScholarCrossref 11. Триго-Гусман
FXConti
AAoki
V
и другие. Приобретенный буллезный эпидермолиз в детском возрасте. J Дерматол 2003; 30
(3)
226-229PubMedGoogle Scholar12.Хорзельский
Т.Карчевская
К.Дыдуч
AKasner
JPieniazek
W Приобретенный буллезный эпидермолиз у мальчика 4 лет. Педиатр дерматол 2000; 17
(2)
157–158PubMedGoogle ScholarCrossref 13. Стюарт
MIWoodley
DTBriggaman
RA Приобретенный буллезный эпидермолиз и связанные с ним симптоматические перепонки пищевода. Arch Dermatol 1991; 127
(3)
373–377PubMedGoogle ScholarCrossref 14.Caux
Ф.Киртшиг
GLemarchand-Venencie
F
и другие.IgA-приобретенный буллезный эпидермолиз у ребенка, приводящий к слепоте. Br J Dermatol 1997; 137
(2)
270-275PubMedGoogle ScholarCrossref 15.Lacour
JPBernard
PRostain
GBaechler-Sadoul
EPisani
AOrtonne
JP В детстве приобрел буллезный эпидермолиз. Педиатр дерматол 1995; 12
(1)
16-20PubMedGoogle ScholarCrossref 16. МакКуэйг
CCChan
LSWoodley
DTRasmussen
JECooper
К.Д. Приобретенный буллезный эпидермолиз в детстве: дифференциация от наследственного буллезного эпидермолиза. Arch Dermatol 1989; 125
(7)
944- 949PubMedGoogle ScholarCrossref 17.Hup
JMBruinsma
РАБОерсма
Эрдэ Йонг
MC Неонатальная пузырчатка обыкновенная: трансплацентарная передача антител. Педиатр дерматол 1986; 3
(6)
468-472PubMedGoogle ScholarCrossref 18. Панко
Дж. Флорелл
SRHadley
JZone
JLeiferman
КВандерхофт
S Неонатальная пузырчатка у ребенка, рожденного от матери с серологическими доказательствами как пузырчатки обыкновенной, так и гестационного пемфигоида. J Am Acad Dermatol 2009; 60
(6)
1057-1062PubMedGoogle ScholarCrossref 19.Bonifazi
EMilioto
МТрашлиева
VFerrante
MRMazzotta
FCoviello
C Вульгарная пузырчатка новорожденных, передающаяся пассивно от матери, у которой отсутствуют клинические симптомы. J Am Acad Dermatol 2006; 55
(5)
(Suppl) S113- S114PubMedGoogle ScholarCrossref 20.Hirsch
Рэндерсон
Дж. Вайнберг
JM
и другие. Неонатальная пузырчатка листовидная. J Am Acad Dermatol 2003; 49
(2)
(дополнительные отчеты о случаях) S187- S189PubMedGoogle ScholarCrossref 22.Lin
MSGharia
MFu
CL
и другие. Молекулярное картирование основных эпитопов ВР180, распознаваемых аутоантителами к герпесу беременности. Clin Immunol 1999; 92
(3)
285-292PubMedGoogle ScholarCrossref 23.Goldberg
NSDeFeo
CКиршенбаум
N Вульгарная пузырчатка и беременность: факторы риска и рекомендации. J Am Acad Dermatol 1993; 28
(5, пт 2)
877-879PubMedGoogle ScholarCrossref 24.Kubo
AHashimoto
Киноуэ
Чашимото
Тёшикава
K Приобретенный буллезный эпидермолиз, усугубляемый системным лечением эстрогенами и прогестероном и беременностью. J Am Acad Dermatol 1997; 36
(5, п.1)
792-794PubMedGoogle ScholarCrossref 25.Kero
MNiemi
К.М.Канерва
L Беременность как триггер приобретенного буллезного эпидермолиза. Acta Derm Venereol 1983; 63
(4)
353-356PubMedGoogle Scholar26.Chowdhury
М.М.Натараджан
S Неонатальная пузырчатка обыкновенная, связанная с легкой вульгарной пузырчаткой полости рта у матери во время беременности. Br J Dermatol 1998; 139
(3)
500-503PubMedGoogle ScholarCrossref
Буллезный эпидермолиз | DermNet NZ
Автор: Ванесса Нган, штатный писатель, 2003 г. Обновлено Джейн Уиддоусон, клиническим медсестрой EB, и др., DEBRA, Новая Зеландия, февраль 2016 г.
Что такое буллезный эпидермолиз?
Буллезный эпидермолиз (БЭ) — это группа наследственных заболеваний, которые характеризуются образованием пузырей на коже и слизистых оболочках. Они могут возникать на любом участке тела, но чаще всего появляются в местах трения и незначительных травм, таких как ступни и руки. В некоторых подтипах волдыри могут также возникать на внутренних органах, таких как пищевод, желудок и дыхательные пути, без видимого трения.
Буллезный эпидермолиз
EB следует отличать от обычных волдырей от трения и от приобретенного буллезного эпидермолиза (EBA), который представляет собой пузырчатое аутоиммунное заболевание, которое не передается по наследству и часто не развивается до взрослой жизни.
Заболевания БЭ возникают в результате генетических дефектов молекул кожи, связанных с адгезией. Потеря адгезии приводит к образованию пузырей. Существует четыре основных типа БЭ, основанных на различных участках образования волдырей в структуре кожи:
Внутри каждого из этих типов БЭ существуют различные подтипы. Каждый тип БЭ имеет разную степень тяжести, от легкой до тяжелой. Международный консенсус в отношении диагностики и классификации БЭ привел к обновленным рекомендациям (2014 г.), основанным на новых клинических и молекулярных данных.
Кто заболевает буллезным эпидермолизом?
EB — это наследственное заболевание, что означает, что вы унаследовали один или два гена EB. При аутосомно-доминантном БЭ для экспрессии болезни необходим только один аномальный ген. Это означает, что только один родитель должен нести ген EB. С другой стороны, аутосомно-рецессивный наследственный EB требует, чтобы у вас было два гена EB (по одному от каждого родителя), чтобы иметь заболевание. Если у человека есть один рецессивный ген EB в паре с нормальным геном, он называется носителем и не болен.
БЭ обычно возникает при рождении или вскоре после него. В равной степени страдают самцы и самки. Иногда БЭ может быть достаточно легкой при рождении, чтобы не проявляться, и обнаруживается только тогда, когда ребенок подрастет или не достигнет совершеннолетия.
Каковы клинические признаки буллезного эпидермолиза?
Простой буллезный эпидермолиз (EBS)
Подробнее см. Простой буллезный эпидермолиз.
| Подтипы EBS | Характеристики |
|---|---|
| Локализованный EBS Ранее известный как Weber-Cockayne |
|
| Generalized EBS Ранее известный как Koebner |
|
| Общая тяжелая EBS Ранее известная как Даулинг Меара |
наблюдается значительное улучшение. |
Буллезный соединительный эпидермолиз (JEB)
Подробнее см. Буллезный соединительный эпидермолиз.
| Подтипы JEB | Характеристики |
|---|---|
| Общая тяжелая форма JEB Ранее известный как Herlitz |
|
| Обобщенный промежуточный JEB Ранее известный как Non-Herlitz |
|
Буллезный дистрофический эпидермолиз (DEB)
Подробнее см. Буллезный дистрофический эпидермолиз.
| Подтипы DEB | Характеристики |
|---|---|
| Доминантный обобщенный DEB |
|
| Генерализованный тяжелый рецессивный (R) DEB Ранее известный как Hallopeau-Siemens; а также; Обобщенный промежуточный RDEB (ранее Non-Hallopeau-Siemens) |
|
Синдром Киндлера
| Синдром Киндлера | Характеристики |
|---|---|
| Синдром Киндлера |
|
Есть много других подтипов EB.На проявление и тяжесть БЭ влияют специфические генетические изменения, которые иногда бывает трудно классифицировать.
Генетическое консультирование и пренатальное тестирование на БЭ доступны в некоторых центрах, и их следует рассмотреть.
Направление за психологической помощью также следует рассматривать для детей с БЭ и их семей, особенно для более тяжелых подтипов.
Как диагностируется буллезный эпидермолиз?
В доминирующих подтипах БЭ, для которых известно информативное генеалогическое древо, часто приемлемо, чтобы клинический диагноз (на основе имеющихся признаков) был поставлен специалистом-дерматологом.
Диагностические тесты также доступны в некоторых странах и включают биопсию кожи недавно образованного волдыря, которая подвергается иммунофлуоресцентному картированию антигенов (IFM) и просвечивающей электронной микроскопии (EM). Мутационный анализ (анализ крови на гены), хотя в настоящее время не считается диагностическим тестом первой линии, также доступен в некоторых странах.
Требуется направление к дерматологу.
Как оценивается степень тяжести буллезного эпидермолиза?
Тяжесть EB можно оценить с помощью следующих систем оценки:
- Оценка Birmingham EB
- EBDASI: Индекс активности БЭ и рубцевания
- iSCOREB: инструмент для оценки клинических результатов исследования EB
- QOLEB: оценка качества жизни в EB
Лечение буллезного эпидермолиза
Общее
Лекарства от БЭ не существует.Однако значительные исследования, включая генную терапию и клеточную терапию, продолжаются с целью улучшения качества жизни.
В большинстве случаев лечение симптоматическое. Основная цель — защитить кожу и остановить образование волдырей, ускорить заживление и предотвратить осложнения.
Поскольку БЭ может поражать очень много различных частей тела, обычно требуется группа медицинских специалистов для оказания общей помощи. При необходимости врач может назначить лечение пероральными и местными лекарствами для облегчения заживления или предотвращения осложнений.
Ниже приведены некоторые общие меры по уходу за пациентом с БЭ.
- Избегайте действий, вызывающих трение кожи. Сюда входит обращение с младенцами и детьми — альтернативным методам обращения с ними легко научиться у квалифицированного специалиста в области здравоохранения.
- Поддерживайте прохладу и избегайте перегрева
- Используйте набивку из пеноматериала или овчину, чтобы уменьшить трение о предметы мебели, такие как кровати, стулья и детские автокресла
- Выбирайте легкую одежду (включая подгузники) и обувь без раздражающих швов и деталей (например, молнии и тугой резинки).
- Прокалывание, дренирование и обработка волдырей для ускорения заживления (это должны делать только люди, прошедшие обучение по уходу за ранами)
- Многие традиционные липкие ленты и повязки могут быть неподходящими для людей с БЭ, особенно с более тяжелыми формами (например, RDEB), поскольку их удаление может вызвать дополнительную травму кожи. Рекомендуется использовать современные продукты для ухода за ранами, такие как силиконовые ленты и повязки с низкой адгезией. Однако находчивость в использовании легкодоступных предметов, таких как нанесение дополнительной смазки (например, вазелин / парафиновое масло) на некоторые традиционные повязки для ран, может оказаться полезной.
- Когда БЭ поражает другие части тела, применяются различные методы ухода и лечения. Например, мягкая диета при поражении пищевода или использование смягчителей стула при запоре, или если у пациента есть анальные волдыри.
- Руководства по клинической практике доступны на веб-сайте DEBRA * International, включая руководства по уходу за полостью рта, уходу за ранами, лечению боли, лечению рака.
- Дополнительная поддержка и информация как от специалистов здравоохранения, так и от лиц, пострадавших от БЭ:
- DEBRA (Исследовательская ассоциация буллезного дистрофического эпидермолиза) насчитывает более 50 групп по всему миру.
- Eb-Clinet — это клиническая сеть центров EB и экспертов по всему миру.
Буллезный эпидермолиз — NHS
Буллезный эпидермолиз (БЭ) — это название группы редких наследственных кожных заболеваний, при которых кожа становится очень хрупкой. Любая травма или трение кожи могут вызвать болезненные волдыри.
Информация:
Консультации по коронавирусу
Получите консультацию по коронавирусу и буллезному эпидермолизу (БЭ) от DEBRA
Симптомы буллезного эпидермолиза
Основные симптомы всех типов БЭ включают:
- кожа, которая легко покрывается волдырями
- волдырей во рту
- волдырей на руках и подошвах ног
- кожа с рубцами, иногда с небольшими белыми пятна, называемые милиами
- утолщение кожи и ногтей
Кредит:
Типы буллезного эпидермолиза
Три основных типа БЭ:
- простой буллезный эпидермолиз (EBS) — наиболее распространенный тип, который имеет тенденцию быть более легким с низким риском серьезных осложнений
- дистрофический буллезный эпидермолиз (DEB) — от легкой до тяжелой
- буллезный узловой эпидермолиз (JEB) — самый редкий и самый тяжелый тип
Тип отражает, где на теле появляются волдыри и какой слой кожи поражен .
Существует также множество вариантов этих трех основных типов БЭ, каждый из которых имеет несколько разные симптомы.
Подробнее о симптомах различных типов буллезного эпидермолиза.
Диагностика EB
EB обычно диагностируется у младенцев и детей вашей неонатальной бригадой, поскольку симптомы часто очевидны с рождения. Но некоторые более легкие типы БЭ могут быть диагностированы только в зрелом возрасте.
Если есть подозрение, что у вашего ребенка такое заболевание, его направят к кожному специалисту (дерматологу).
Специалист проведет обследование для определения типа БЭ и поможет составить план лечения. Они могут взять небольшой образец кожи (биопсия) для отправки на исследование.
Пренатальное тестирование
В некоторых случаях можно проверить еще не родившегося ребенка на БЭ примерно на 11 неделе беременности.
Это может быть предложено, если известно, что вы или ваш партнер являетесь носителем дефектного гена, связанного с БЭ, и есть риск рождения ребенка с тяжелым типом БЭ.
Если тест подтвердит, что у вашего ребенка БЭ, вам будут предложены консультации и рекомендации, которые помогут вам принять обоснованное решение о том, как вы хотите продолжить беременность.
Пренатальные тесты включают амниоцентез и забор проб ворсин хориона.
Причины буллезного эпидермолиза
EB вызывается дефектным геном (генная мутация), который делает кожу более хрупкой.
Обычно ребенок с EB унаследует дефектный ген от родителя, у которого также есть EB.
Также возможно, что ребенок с EB унаследовал дефектный ген от обоих родителей, которые являются просто «носителями», но сами не имеют EB.
Лечение буллезного эпидермолиза
В настоящее время лекарства от БЭ не существует, поэтому лечение направлено на облегчение симптомов и предотвращение развития осложнений, таких как инфекция.
Группа медицинских специалистов поможет вам решить, какое лечение лучше всего подходит для вашего ребенка, и посоветует, как жить с этим заболеванием.
Вы можете лечить БЭ в домашних условиях:
- лопать волдыри стерильной иглой
- накладывать защитные повязки
- избегать вещей, ухудшающих состояние
Лекарства можно использовать для лечения инфекции или уменьшения боли. Операция может потребоваться, если БЭ вызывает сужение пищевода или проблемы с руками.
Подробнее о лечении буллезного эпидермолиза.
Приобретенный буллезный эпидермолиз
Приобретенный буллезный эпидермолиз (ББЭ) — приобретенная форма БЭ с аналогичными симптомами.
Как и EB, EBA легко вызывает образование волдырей. Это также может повлиять на ротовую полость, горло и пищеварительный тракт.
Но EBA не передается по наследству, и симптомы обычно не проявляются до более позднего возраста.
Это аутоиммунное заболевание, которое означает, что ваша иммунная система начинает атаковать здоровые ткани тела. Точно не известно, что вызывает это.
EBA — очень редкое заболевание, которое чаще встречается у людей старше 40 лет.
Благотворительные организации и группы поддержки
Если вашему ребенку поставили диагноз БЭ, это может быть пугающим и подавляющим опытом.Вероятно, вы захотите узнать как можно больше о состоянии и доступных методах лечения.
DEBRA — это национальная благотворительная организация, которая предоставляет помощь, советы и поддержку людям в Великобритании, живущим с EB.
DEBRA International — это всемирная сеть национальных групп, работающих от имени людей, пострадавших от БЭ.
Поддержка лиц, осуществляющих уход
При уходе за ребенком со сложным и тяжелым заболеванием, таким как БЭ, важно не пренебрегать своим здоровьем и благополучием.
Прочтите о перерывах в уходе и временном уходе и получите советы по уходу за ребенком-инвалидом.
Информация о вас
Если у вас или вашего ребенка есть EB, ваша клиническая бригада передаст информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Подробнее о реестре
Последняя проверка страницы: 19 марта 2018 г.
Срок следующей проверки: 19 марта 2021 г.
Буллезный эпидермолиз — NORD (Национальная организация по редким заболеваниям)
ТЕКСТОВЫЕ КНИГИ
Fine JD, Hintner H., eds. Жизнь с буллезным эпидермолизом (БЭ): этиология, диагностика, многопрофильная помощь и терапия, Нью-Йорк: Springer Wien; 2009.
Fine JD, Bauer E, McGuire J, Moshell A, eds. Буллезный эпидермолиз: клинические, эпидемиологические и лабораторные достижения и результаты Национального реестра буллезного эпидермолиза 1-е издание The Johns Hopkins University Press; 1999 г.
Weston WL, Lane AT, Morelli JG, ред. Цветной учебник детской дерматологии, 4-е издание Mosby / Elsevier, 2007: 348-354.
ОБЗОР СТАТЬИ
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть I — Патогенез и клинические особенности. Дерматологические клиники, Том 28/1. Филадельфия: W.B. Saunders Elsevier.
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть II — Диагностика и лечение. Дерматологические клиники, Том 28/2. Филадельфия: W.B. Saunders Elsevier.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть I. Ткани, ассоциированные с эпителием.
J Am Acad Dermatol. 2009 сентябрь; 61 (3): 367-84.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть II. Другие органы. J Am Acad Dermatol. 2009 сентябрь; 61 (3): 387-402.
Pfendne EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Фундаментальная наука о буллезном эпидермолизе, его диагностическая и молекулярная характеристика: Материалы II Международного симпозиума по буллезному эпидермолизу.Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 781-794.
Азизхан Р.Г., Дениер Дж.Э., Меллерио Дж.Э., Гонсалес Р., Бачигалупо М., Кантор А., Пассалаква Г., Палиссон Ф., Лаки А.В. Хирургическое лечение буллезного эпидермолиза: материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 г. Международный журнал дерматологии. 2007: 46 (8): 801-808.
Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, Palisson F. Медицинское лечение буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии 2007; 46 (8): 795-800.
Lucky AW, Pfendner E, Pillay E, Paskel J, Weiner M, Palisson F. Психосоциальные аспекты буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 809-814.
ИНТЕРНЕТ
Debra International. Уход за больным БЭ. Доступно по адресу: http://www.debra-international.org/patients/caring-for-someone-with-eb.html По состоянию на 30 мая 2013 г.
Маринкович М.П.Буллезный эпидермолиз Обновлено: 10 августа 2012 г. Medscape. Доступно по адресу: http://emedicine.medscape.com/article/1062939-overview По состоянию на 30 мая 2013 г.
Пфенднер Э., Брукнер А. Простой буллезный эпидермолиз. Последнее обновление 1 сентября 2011 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Дистрофический буллезный эпидермолиз.Последнее обновление 4 ноября 2010 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный узловой эпидермолиз. Размещено: 2 февраля 2008 г. В: Обзоры генов на сайте GeneTests: информационный ресурс по медицинской генетике (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно по адресу http: //www.genetests.орг. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный эпидермолиз с атрезией привратника. Последнее обновление: 14 февраля 2013 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно на http://www.genetests.org По состоянию на 30 мая 2013 г.
Дистрофический буллезный эпидермолиз: MedlinePlus Genetics
Дистрофический буллезный эпидермолиз — одна из основных форм группы состояний, называемых буллезным эпидермолизом.Буллезный эпидермолиз делает кожу очень хрупкой и легко покрывается волдырями. Волдыри и эрозии кожи образуются в результате незначительной травмы или трения, например, трения или царапин. Признаки и симптомы дистрофического буллезного эпидермолиза широко различаются у разных людей. В легких случаях образование волдырей может в первую очередь поражать руки, ступни, колени и локти. Тяжелые случаи этого состояния включают широко распространенные волдыри, которые могут привести к потере зрения, рубцам и другим серьезным проблемам со здоровьем.
Исследователи классифицируют дистрофический буллезный эпидермолиз на основные типы в зависимости от характера наследования и особенностей заболевания. Хотя типы различаются по степени тяжести, их особенности значительно перекрываются, и они вызваны мутациями в одном и том же гене.
Рецессивный генерализованный дистрофический буллезный эпидермолиз тяжелой степени (RDEB-sev gen) является классической формой заболевания и является наиболее тяжелой. Заболевшие младенцы обычно рождаются с широко распространенными волдырями и участками без кожи, часто вызванными травмой, полученной во время родов.Чаще всего волдыри присутствуют по всему телу и поражают слизистые оболочки, такие как влажная слизистая рта и пищеварительного тракта. По мере заживления волдырей образуются серьезные рубцы. Рубцы во рту и пищеводе могут затруднить пережевывание и проглатывание пищи, что приводит к хроническому недоеданию и замедлению роста. Дополнительные осложнения продолжающегося рубцевания могут включать сращение кожи между пальцами рук и ног, потерю ногтей на руках и ногах, деформации суставов (контрактуры), ограничивающие движение, и воспаление глаз, ведущее к потере зрения.Кроме того, люди с RDEB-sev gen имеют очень высокий риск развития в молодом возрасте формы рака кожи, называемой плоскоклеточной карциномой. У этих людей рак имеет тенденцию быть необычно агрессивным и часто опасным для жизни.
Другие типы рецессивного дистрофического буллезного эпидермолиза относятся к спектру, который называется генерализованным и локализованным RDEB (RDEB-gen и -loc). Эти формы состояния несколько менее тяжелы, чем RDEB-sevgen, и различаются по пораженным участкам тела.Волдыри часто ограничиваются кистями, стопами, коленями и локтями в легких случаях, но могут быть широко распространенными в более тяжелых случаях. Редкие формы поражают определенные участки тела, такие как голени или живот. У больных часто бывают деформированные ногти на руках и ногах. Типы RDEB-gen и -loc включают рубцевание в областях, где возникают волдыри, но эти формы состояния не вызывают серьезных рубцов, характерных для RDEB-sev gen.
Другой важный тип этого состояния известен как доминирующий дистрофический буллезный эпидермолиз (ДДЭБ).Признаки и симптомы этого состояния, как правило, более легкие, чем у рецессивных форм, с волдырями, которые часто ограничиваются руками, ступнями, коленями и локтями. Волдыри заживают рубцами, но это менее серьезно, чем при рецессивных формах этого состояния. У большинства пораженных людей ногти на руках и ногах деформированы, и со временем ногти могут выпадать. В самых легких случаях аномальные ногти — единственный признак этого состояния.
Детский буллезный эпидермолиз: история болезни, патофизиология, эпидемиология
Автор
Сурасак Пувабандицин, доктор медицины Доцент педиатрии, Медицинская школа Рутгерса Роберта Вуда Джонсона; Адъюнкт-профессор педиатрии медицинского факультета Университета Святого Георгия; Адъюнкт-профессор педиатрии Школы последипломного медицинского образования Университета Сетон Холл; Доцент кафедры педиатрии, Медицинская школа Рутгерса, Нью-Джерси
Сурасак Пувабандицин, доктор медицинских наук, является членом следующих медицинских обществ: Американская академия педиатрии
Раскрытие информации: нечего раскрывать.
Соавтор (ы)
Раджив Мехта, доктор медицины, FRCP Профессор кафедры педиатрии Медицинской школы Рутгерса Роберта Вуда Джонсона; Заместитель директора по неонатологии и содиректор прикладных исследований (педиатрия), Детская больница Бристол-Майерс Сквибб
Раджив Мехта, доктор медицины, FRCP является членом следующих медицинских обществ: Восточного общества педиатрических исследований, Королевского колледжа врачей Ирландии , Королевский колледж врачей и хирургов Соединенного Королевства
Раскрытие информации: раскрывать нечего.
Дженнифер Ай МакКоннелл, доктор медицины Врач-резидент, отделение педиатрии, Медицинская школа Рутгерса Роберта Вуда Джонсона
Дженнифер Ай МакКоннелл, доктор медицины, является членом следующих медицинских обществ: Американская академия педиатрии
Раскрытие информации: раскрывать нечего.
Натали Кей Генгель, DO Врач-резидент, Отделение педиатрии, Медицинская школа Рутгерса Роберта Вуда Джонсона
Натали Кей Генгель, DO является членом следующих медицинских обществ: Американская академия педиатрии
Раскрытие информации: раскрывать нечего.
Джулия Мэйн, MD Врач-резидент, Отделение педиатрии, Медицинская школа Рутгерса Роберта Вуда Джонсона
Раскрытие: Ничего не разглашать.
Lauren Walzer, DO Врач-резидент, отделение педиатрии, детская больница Bristol-Myers Squibb, медицинская школа Рутгерса Роберта Вуда Джонсона
Lauren Walzer, DO является членом следующих медицинских обществ: Американская академия педиатрии
Раскрытие информации: Нечего раскрывать.
Специальная редакционная коллегия
Мэри Л. Виндл, PharmD Адъюнкт-профессор, Фармацевтический колледж Медицинского центра Университета Небраски; Главный редактор Medscape Drug Reference
Раскрытие информации: нечего раскрывать.
Роберт А. Шварц, доктор медицины, магистр здравоохранения Профессор и руководитель дерматологии, профессор патологии, профессор педиатрии, профессор медицины, Медицинская школа Рутгерса, Нью-Джерси
Роберт А. Шварц, доктор медицины, магистр здравоохранения является членом следующих медицинских обществ : Alpha Omega Alpha, Американская академия дерматологии, Нью-Йоркская медицинская академия, Королевский колледж врачей Эдинбурга, Sigma Xi
Раскрытие: Ничего не разглашать.
Главный редактор
Дирк Элстон, доктор медицины Профессор и председатель кафедры дерматологии и дерматологической хирургии Медицинского университета Южной Каролины Медицинский колледж
Дирк Элстон, доктор медицины, является членом следующих медицинских обществ: Американская академия дерматологии
Раскрытие информации : Нечего раскрывать.
Дополнительные участники
Кевин П. Коннелли, DO доцент кафедры педиатрии, отделение общей педиатрии и неотложной помощи, Медицинский факультет Университета Содружества Вирджинии; Медицинский директор программы посещения домашних животных «Лапы за здоровье» Ричмондской ассоциации охраны здоровья и животных; Педиатрический врач скорой помощи, Emergency Consultants Inc., Медицинский центр Чиппенхэма
Кевин П. Коннелли, DO, является членом следующих медицинских обществ: Американской академии педиатрии, Американского колледжа педиатров-остеопатов, Американской остеопатической ассоциации
Раскрытие информации: раскрывать нечего.
Alexis A D’Elia, MD Сотрудник по сердечно-сосудистым заболеваниям, Университетская больница Уинтропа; Помощник клинического инструктора, Государственный университет Нью-Йорка в Медицинской школе Стоуни-Брук
Алексис А. Д’Элия, доктор медицины, является членом следующих медицинских обществ: Американский колледж врачей
Раскрытие информации: не подлежит разглашению.
Rungtiwa Weerasethsiri, MD Главный педиатр, Merced Facuty Associates, California
Rungtiwa Weerasethsiri, MD является членом следующих медицинских обществ: Американская академия педиатрии
Раскрытие информации: раскрывать нечего.
Nisha Patel, MD Врач-резидент, Westchester Medical Center, Детская больница Maria Ferrari
Nisha Patel, MD является членом следующих медицинских обществ: Американская ассоциация студентов-медиков / Фонд Phi Beta Kappa
Раскрытие информации: раскрытие информации .
Эрик Брандсма, доктор медицинских наук, научный сотрудник, Отделение неонатологии, Университет медицины и стоматологии Нью-Джерси
Раскрытие: Ничего не разглашать.
Благодарности
Авторы благодарят Джуди Уилкинсон, библиотекаря Медицинского центра Джерси-Сити, за ее помощь. Авторы также благодарят Сильвию Саттон-Торп, Кристал Пувабандистин и Кристину Пувабандицин за поддержку этих усилий и подготовку рукописи.
Авторы и редакторы Medscape Drugs & Diseases с благодарностью признают вклад предыдущего соавтора Юджина Гарроу, доктора медицины, в первоначальное написание и разработку этой статьи.
Генетическое тестирование на врожденный буллезный эпидермолиз
Биоинформатика
Целевая область для каждого гена включает кодирующие экзоны и ± 20 пар оснований от границы экзон-интрон. Кроме того, панель включает варианты без кодирования и нормативные требования, если они перечислены выше (варианты без кодирования, охватываемые панелью). Некоторые участки гена (ов) могут быть удалены из панели, если это специально указано в разделе «Ограничения теста» выше. Если тест включает митохондриальный геном, список генов целевой области содержит митохондриальные гены.Данные секвенирования, полученные в нашей лаборатории, анализируются с помощью нашего запатентованного конвейера анализа данных и аннотаций, объединяющего самые современные алгоритмы и стандартные программные решения. Включение строгих шагов контроля качества на протяжении всего рабочего процесса конвейера обеспечивает согласованность, достоверность и точность результатов. Наш конвейер оптимизирован, чтобы обеспечить максимальную чувствительность без ущерба для специфичности. Мы включили ряд баз данных эталонных популяций и баз данных мутаций, включая, помимо прочего, 1000 Genomes Project, gnomAD, ClinVar и HGMD в наше программное обеспечение для клинической интерпретации, чтобы сделать процесс эффективным и действенным.Для вариантов с отсутствием смысла, in silico инструменты прогнозирования вариантов , такие как SIFT, PolyPhen,
MutationTaster используются для помощи в классификации вариантов. Через нашу онлайн-систему заказов и отчетов, Nucleus, поставщики заказов имеют доступ к деталям анализа, включая метрики секвенирования для конкретного пациента, график уровня охвата генов и список регионов с субоптимальным охватом (<20X для ядерных генов и <1000X для мтДНК), если применимо. Это отражает нашу миссию по созданию полностью прозрачной диагностики, при которой поставщики услуг могут легко визуализировать важные детали процесса анализа.
Клиническая интерпретация
Мы предоставляем клиентам самый полный клинический отчет, доступный на рынке. Клиническая интерпретация требует фундаментального понимания клинической генетики и генетических принципов. В Blueprint Genetics наши кандидаты наук по молекулярной генетике, медицинские генетики и клинические консультанты вместе готовят клиническое заключение, оценивая идентифицированные варианты в контексте фенотипической информации, предоставленной в форме заявки.Наша цель — предоставить клинически значимые утверждения, понятные для всех медицинских работников, независимо от того, имеют ли они формальное образование в области генетики.
Классификация вариантов является краеугольным камнем клинической интерпретации и вытекающих из нее решений по ведению пациентов. Наши классификации соответствуют рекомендациям ACMG 2015.
Последний шаг в анализе — ортогональное подтверждение. Варианты последовательности и числа копий, классифицируемые как патогенные, вероятные патогенные и варианты с неопределенной значимостью (VUS), подтверждаются с помощью двунаправленного секвенирования по Сэнгеру ортогональными методами, такими как qPCR / ddPCR, когда они не соответствуют нашим строгим показателям качества NGS для истинно положительного результата. .
Наше клиническое заявление включает таблицы для секвенирования и вариантов числа копий, которые включают основную информацию о вариантах (геномные координаты, номенклатура HGVS, зиготность, частоты аллелей, предсказания in silico, фенотипы OMIM и классификация варианта). Кроме того, заявление включает подробное описание варианта, гена и фенотипа (ов), включая роль конкретного гена в заболевании человека, профиль мутации, информацию об изменении гена в популяционных когортах и подробную информацию о связанных фенотипах.Мы также предоставляем ссылки на справочные материалы, рефераты и базы данных вариантов, которые используются, чтобы помочь поставщикам заказов в дальнейшей оценке полученных результатов, если это необходимо. Заключение суммирует всю имеющуюся информацию и дает наше обоснование классификации варианта.
Выявление патогенных или вероятных патогенных вариантов при доминантных расстройствах или их комбинаций в различных аллелях при рецессивных расстройствах считается молекулярным подтверждением клинического диагноза.В этих случаях для стратификации риска можно использовать тестирование членов семьи. Мы не рекомендуем использовать варианты с неопределенной значимостью (VUS) для стратификации риска членов семьи или ведения пациентов. Рекомендуется генетическое консультирование.
Наша команда интерпретаторов анализирует миллионы вариантов от тысяч людей с редкими заболеваниями. Наша внутренняя база данных и наше понимание вариантов и родственных фенотипов увеличивается с каждым анализируемым случаем. Таким образом, наша лаборатория имеет все возможности для переклассификации ранее описанных вариантов по мере появления новой информации.Если вариант, о котором ранее сообщала Blueprint Genetics, переклассифицируется, наша лаборатория без дополнительных затрат выдаст последующее заявление первому заказавшему его поставщику медицинских услуг.
.