
Скрининг из пятки новорожденного: Для чего нужен неонатальный скрининг?

Newborn Screening — Russia
Неонатальный скрининг от Igenomix – это комплексный генетический тест, с помощью которого анализируются 237 генов методом NGS.
Заинтересовались?
Узнать больше
Наш email: [email protected]
Общая информация
- Неонатальный скрининг
- Преимущества
- Показания
Общая информация
- Около 3% -4% новорожденных имеют генетическое заболевание.
- Неонатальный скрининг – это пункт обязательной программы общественного здравоохранения, который обеспечивает исследования и последующее медицинское обслуживание при различных заболеваниях.
- Скрининговый тест Igenomix для новорожденных (IGX-NBS) – это комплексный генетический тест, с помщью которого анализируются 237 генов с использованием технологии секвенирования нового поколения (NGS), позволяющая напрямую определять генетические нарушения для быстрой и точной диагностики.

- Кроме того, этот тест определяет, является ли ребенок здоровым носителем любого из входящих в панель заболеваний.

Неонатальный скрининг от Igenomix предоставляет расширенную панель заболеваний, проанализированных с помощью технологий на основе NGS, предлагающих более широкий охват, чем подобные стандартные скрининги.
- Эти гены отвечают за нарушения развития, генетические и метаболические нарушения, которые вызывают серьезные проблемы со здоровьем, начиная с раннего детства.
- Главным преимуществом является раннее вмешательство с первого дня жизни для предотвращения умственной и физической инвалидизации, а также развития опасных для жизни заболеваний.
- Выявляется гораздо больше заболеваний, чем при обычном анализе крови.
Узнать больше
Цели обычного скрининга новорожденных
Цели скрининга новорожденных:
- Снижение заболеваемости и смертности от болезней, поддающихся лечению, путем проведения раннего вмешательства для улучшения неонатальных и долгосрочных результатов в отношении здоровья.

- Обеспечить всеобщее медицинское обследование всех новорожденных.
- Выявление новорожденных с положительным результатом скрининга.
- Диагностика заболеваний и состояний.
- Общение с семьями
- Направление в лечебные центры
- Долгосрочное наблюдение.
- Обучение врачей и пациентов.
Процесс проведения исследования?
Ограничения исследования
- Скрининг новорожденных НЕ заменяет генетическое тестированин на носительство моногенных заболеваний (CGT), а генетическое тестирование на носительство не заменяет скрининг новорожденных.
- Выполняется в первые дни жизни.
- Необходим образец крови (анализ слюны ожидает утверждения в Igenomix)
Критерии и классификация
Что было включено и почему?
Заболевания, включенные в скрининг новорожденных, были выбраны по следующим критериям:
- Заболевания с высокой распространенностью.
- Выявление болезней, при которых можно принять меры для улучшения прогноза.

- Отбор генов по данным доказательной медицины.
- Польза скрининга должна перевышать риски.
- Возможность скрининга для населения в целом на местном и национальном уровне.
- Наличие и доступ к лечению.
- Обучение врачей и повышение осведомленности общественности.
Скрининг новорожденных Igenomix по сравнению с текущими неонатальным скринингом
Наш неонатальный скрининг включает в себя все болезни из стандартного скрининга новорожденных (кровь из пятки новорожденного), а также из Рекомендованной унифицированной скрининговой панели (RUSP) и многое другое:
Заболевания, включенные в неонатальный скрининг от Igenomix, можно разделить на следующие группы. Вы можете найти пример болезни, включенной в каждую группу, и ее лечения.
Группа заболевания | Заболевание | Сиптомы | Лечение | Результат |
Заболевания обмена веществ | Синдром Менкеса | При отсутствии лечения характеризуется трудностями при кормлении, вялостью, рвотой, запахом кленового сиропа в ушной сере и моче, энцефалопатией и центральной дыхательной недостаточностью. | Терапия с помощью назначения диеты для снижения токсичных метаболитов и достижения необходимых концентраций недостающих аминокислот в плазме крови. | Бессимптомная нормальная жизнь |
Иммунодефициты | Тяжелый комбинированный иммунодефицит (ТКИД)
| Рецидивирующие, тяжело протекающие инфекции, вызванные условно-патогенными микроорганизмами, задержка развития | Защитные меры профилактики с помощью антибиотиков, противогрибковых, противовирусных препаратов и замены антител. | Снижение частоты инфекций, замедление прогрессирования заболевания и улучшение прогноза. |
Неврология | Болезнь Вильсона | Боли в животе, желтуха, гепатоспленомегалия, асцит, кровотечение из верхних отделов желудочно-кишечного тракта и поражение нервной системы. | Раннее начало хелатирования меди | Уменьшение накопления меди и предотвращение необратимых повреждений. |
Пульмонология | Муковисцидоз | Новорожденные страдают выпадением прямой кишки, мекониевой непроходимостью, респираторными инфекциями, недостаточностью поджелудочной железы, а также другими симптомами. | Раннее начало легочной терапии, диетотерапии, антибиотикопрофилактики, вакцинации, бронходилататоров. | Уменьшаетстя частота инфекций, поддерживается функция легких. Уменьшение прогрессирования заболевания и улучшение прогноза. |
Эндокринология | Врожденный гипотиреоз | У большинства младенцев болезнь протекает бессимптомно, у детей с симптомами обычно наблюдаются летаргия, хриплый крик, проблемы с приемом пищи, запоры, микседема, макроглоссия и другие симптомы. | Раннее начало заместительной терапии гормонов щитовидной железы (пероральный левотироксин) | Предотвращение развития нейрокогнитивных нарушений (измеряется коэффициеном IQ) |
Отоларингология | Наследственная тугоухость* | Новорожденные, скорее всего, не будут иметь симптомов до раннего детства, когда пациенту станет сложно понимать слова, слышать согласные и т. | Раннее начало использования слуховых аппаратов, логопеда и языковой терапии | Предотвращение задержки развития и речи |
Гемоглобинопатии | Серповидноклеточная анемия | Новорожденные могут иметь тяжелую анемию, вазоокклюзивный криз, хроническую боль в животе, гипоспленизм и инфекционные заболевания. | Раннее начало первичной профилактики острых осложнений, а также трансплантация гемопоэтических стволовых клеток | Предотвращение рецидивирующих острых вазоокклюзивных эпизодов и в сочетании с успешной трансплантацией гемопоэтических стволовых клеток добиться нормальной бессимптомной жизни. |
Нервно-мышечные заболевания | Спинальная мышечная атрофия | Характеризуется диффузной симметричной проксимальной мышечной слабостью, более выраженной в нижних конечностях, с отсутствием или заметно сниженными глубокими сухожильными рефлексами. | Раннее начало терапии, модифицирующей заболевание, и поддерживающей терапии. | Улучшение качества жизни и увеличение продолжительности жизни. |


 д.
д.
Все гены и заболевания
| ГЕН | ЗАБОЛЕВАНИЕ (OMIM) | ТИП ЗАБОЛЕВАНИЯ | ТИП НАСЛЕДОВАНИЯ |
| ABCD1 | Adrenoleukodystrophy | Metabolic disorder- Fatty acid oxidation | XL |
| ABCD4 | Methylmalonic aciduria and homocystinuria, cblJ type | Metabolic disorder- Organic acid | AR |
| ACAD8 | Isobutyryl-CoA dehydrogenase deficiency | Metabolic disorder- Organic acid | AR |
| ACADM | Acyl-CoA dehydrogenase, medium chain, deficiency of | Metabolic disorder- Fatty acid oxidation | AR |
| ACADS | Acyl-CoA dehydrogenase, short-chain, deficiency of | Metabolic disorder- Fatty acid oxidation | AR |
| ACADSB | 2-methylbutyrylglycinuria | Metabolic disorder- Organic acid | AR |
| ACADVL | VLCAD deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| ACAT1 | Alpha-methylacetoacetic aciduria | Metabolic disorder- Organic acid | AR |
| ADA | Severe combined immunodeficiency due to ADA deficiency | Immunodeficiency disorder | AR |
| ADK | Hypermethioninemia due to adenosine kinase deficiency | Metabolic disorder- Amino acid | AR |
| AHCY | Hypermethioninemia with deficiency of S-adenosylhomocysteine hydrolase | Metabolic disorder- Amino acid | AR |
| ALDh5A1 | Hyperprolinemia, type II | Metabolic disorder- Amino acid | AR |
| AMT | Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| ARG1 | Argininemia | Metabolic disorder- Amino acid | AR |
| ASL | Argininosuccinic aciduria | Metabolic disorder- Amino acid | AR |
| ASS1 | Citrullinemia | Metabolic disorder- Amino acid | AR |
| ATP7B | Wilson disease | Metabolic disorder- Copper | AR |
| AUH | 3-methylglutaconic aciduria, type I | Metabolic disorder- Organic acid | AR |
| BCKDHA | Maple syrup urine disease, type Ia | Metabolic disorder- Amino acid | AR |
| BCKDHB | Maple syrup urine disease, type Ib | Metabolic disorder- Amino acid | AR |
| BTD | Biotinidase deficiency | Metabolic disorder- Amino acid | AR |
| CBS | Homocystinuria, B6-responsive and nonresponsive types | Metabolic disorder- Amino acid | AR |
| CFTR | Cystic fibrosis | Respiratory disorder | AR |
| CPT1A | CPT deficiency, hepatic, type IA | Metabolic disorder- Fatty acid oxidation | AR |
| CPT2 | CPT II deficiency, lethal neonatal | Metabolic disorder- Fatty acid oxidation | AR |
| CTH | Cystathioninuria | Metabolic disorder- Amino acid | AR |
| CYP11B1 | Adrenal hyperplasia, congenital, due to 11-beta-hydroxylase deficiency | Endocrine disorder | AR |
| CYP21A2 | Adrenal hyperplasia, congenital, due to 21-hydroxylase deficiency | Endocrine disorder | AR |
| DBT | Maple syrup urine disease, type II | Metabolic disorder- Amino acid | AR |
| DNAJC12 | Hyperphenylalaninemia, mild, non-Bh5-deficient | Metabolic disorder- Amino acid | AR |
| DUOX2 | Thyroid dyshormonogenesis 6 | Endocrine disorder | AR |
| DUOXA2 | Thyroid dyshormonogenesis 5 | Endocrine disorder | AR |
| ETFA | Glutaric acidemia IIA | Metabolic disorder- Organic acid | AR |
| ETFB | Glutaric acidemia IIB | Metabolic disorder- Organic acid | AR |
| ETFDH | Glutaric acidemia IIC | Metabolic disorder- Organic acid | AR |
| FAH | Tyrosinemia, type I | Metabolic disorder- Amino acid | AR |
| GAA | Glycogen storage disease II | Metabolic disorder- Glicogen storage | AR |
| GALE | Galactose epimerase deficiency | Metabolic disorder- Carbohydrate | AR |
| GALK1 | Galactokinase deficiency with cataracts | Metabolic disorder- Carbohydrate | AR |
| GALT | Galactosemia | Metabolic disorder- Carbohydrate | AR |
| GCDH | Glutaric acidemia, type I | Metabolic disorder- Organic acid | AR |
| GCh2 | Hyperphenylalaninemia, Bh5-deficient, B | Metabolic disorder- Amino acid | AR |
| GCSH | ?Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| GJB2 | Deafness, digenic GJB2/GJB6 | Auditory disorder | AR |
| GJB6 | Deafness, digenic GJB2/GJB6 | Auditory disorder | AR |
| GLDC | Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| GNMT | Glycine N-methyltransferase deficiency | Metabolic disorder- Amino acid | AR |
| GSS | Glutathione synthetase deficiency | Metabolic disorder- Organic acid | AR |
| HADH | 3-hydroxyacyl-CoA dehydrogenase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HADHA | LCHAD deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HADHB | Trifunctional protein deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HBB | Sickle cell anemia | Hemoglobin disorder | AR |
| HCFC1 | Mental retardation, X-linked 3 (methylmalonic acidemia and homocysteinemia, cblX type ) | Metabolic disorder- Organic acid | XLR |
| HGD | Alkaptonuria | Metabolic disorder- Amino acid | AR |
| HLCS | Holocarboxylase synthetase deficiency | Metabolic disorder- Organic acid | AR |
| HMGCL | HMG-CoA lyase deficiency | Metabolic disorder- Organic acid | AR |
| HPD | Tyrosinemia, type III | Metabolic disorder- Amino acid | AR |
| HSD17B10 | HSD10 mitochondrial disease | Metabolic disorder- Organic acid | XLD |
| IDUA | Mucopolysaccharidosis Ih/s | Metabolic disorder- Lysosomal enzime | AR |
| IL2RG | Severe combined immunodeficiency, X-linked | Immunodeficiency disorder | XLR |
| IVD | Isovaleric acidemia | Metabolic disorder- Organic acid | AR |
| LMBRD1 | Methylmalonic aciduria and homocystinuria, cblF type | Metabolic disorder- Organic acid | AR |
| MAT1A | Methionine adenosyltransferase deficiency, autosomal recessive | Metabolic disorder- Amino acid | AR |
| MCCC1 | 3-Methylcrotonyl-CoA carboxylase 1 deficiency | Metabolic disorder- Organic acid | AR |
| MCCC2 | 3-Methylcrotonyl-CoA carboxylase 2, deficiency | Metabolic disorder- Organic acid | AR |
| MCEE | Methylmalonyl-CoA epimerase deficiency | Metabolic disorder- Organic acid | AR |
| MLYCD | Malonyl-CoA decarboxylase deficiency | Metabolic disorder- Organic acid | AR |
| MMAA | Methylmalonic aciduria, vitamin B12-responsive | Metabolic disorder- Organic acid | AR |
| MMAB | Methylmalonic aciduria, vitamin B12-responsive, due to defect in synthesis of adenosylcobalamin, cblB complementation type | Metabolic disorder- Organic acid | AR |
| MMACHC | Methylmalonic aciduria and homocystinuria, cblC type | Metabolic disorder- Organic acid | AR |
| MMADHC | Methylmalonic aciduria and homocystinuria, cblD type | Metabolic disorder- Organic acid | AR |
| MMUT | Methylmalonic aciduria, mut(0) type | Metabolic disorder- Organic acid | AR |
| MTHFR | Homocystinuria due to MTHFR deficiency | Metabolic disorder- Amino acid | AR |
| MTR | Homocystinuria-megaloblastic anemia, cbl E type | Metabolic disorder- Amino acid | AR |
| MTRR | Homocystinuria due to MTHFR deficiency | Metabolic disorder- Amino acid | AR |
| MVK | Mevalonic aciduria | Metabolic disorder- Organic acid | AR |
| NADK2 | ?2,4-dienoyl-CoA reductase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| OTC | Ornithine transcarbamylase deficiency | Metabolic disorder- Amino acid | XLR |
| PAH | Phenylketonuria | Metabolic disorder- Amino acid | AR |
| PAX8 | Hypothyroidism, congenital, due to thyroid dysgenesis or hypoplasia | Endocrine disorder | AD |
| PCBD1 | Hyperphenylalaninemia, Bh5-deficient, D | Metabolic disorder- Amino acid | AR |
| PCCA | Propionic acidemia | Metabolic disorder- Organic acid | AR |
| PCCB | Propionic acidemia | Metabolic disorder- Organic acid | AR |
| PPM1K | ?Maple syrup urine disease, mild variant | Metabolic disorder- Organic acid | AR |
| PRDX1 | Methylmalonic aciduria and homocystinuria, cblC type, digenic | Metabolic disorder- Organic acid | AR |
| PRODH | Hyperprolinemia, type I | Metabolic disorder- Amino acid | AR |
| PTS | Hyperphenylalaninemia, Bh5-deficient, A | Metabolic disorder- Amino acid | AR |
| QDPR | Hyperphenylalaninemia, Bh5-deficient, C | Metabolic disorder- Amino acid | AR |
| RAG1 | Severe combined immunodeficiency, B cell-negative | Immunodeficiency disorder | AR |
| RAG2 | Severe combined immunodeficiency, B cell-negative | Immunodeficiency disorder | AR |
| SLC22A5 | Carnitine deficiency, systemic primary | Metabolic disorder- Fatty acid oxidation | AR |
| SLC25A13 | Citrullinemia, adult-onset type II | Metabolic disorder- Amino acid | AR |
| SLC25A20 | Carnitine-acylcarnitine translocase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| SLC3A1 | Cystinuria | Metabolic disorder- Amino acid | AR |
| SLC5A5 | Thyroid dyshormonogenesis 1 | Endocrine disorder | AR |
| SLC6A8 | Cerebral creatine deficiency syndrome 1 | Metabolic disorder- Fatty acid oxidation | XLR |
| SLC7A7 | Lysinuric protein intolerance | Metabolic disorder- Amino acid | AR |
| SLC7A9 | Cystinuria | Metabolic disorder- Amino acid | AR |
| SMN1 | Spinal muscular atrophy | Neuromuscular disorder | AR |
| TAT | Tyrosinemia, type II | Metabolic disorder- Amino acid | AR |
| TG | Thyroid dyshormonogenesis 3 | Endocrine disorder | AR |
| TPO | Thyroid dyshormonogenesis 2A | Endocrine disorder | AR |
| TSHB | Hypothyroidism, congenital, nongoitrous 4 | Endocrine disorder | AR |
| TSHR | Hypothyroidism, congenital, nongoitrous, 1 | Endocrine disorder | AR |
Ссылки на публикации
Связанные публикации:
ACOG Committee Opinion No. 778 Summary: Newborn Screening and the Role of the Obstetrician–Gynecologist. (2019). Obstetrics & Gynecology, 133(5), 1073-1074. doi: 10.1097/aog.0000000000003246
778 Summary: Newborn Screening and the Role of the Obstetrician–Gynecologist. (2019). Obstetrics & Gynecology, 133(5), 1073-1074. doi: 10.1097/aog.0000000000003246
https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html#:~:text=The%20RUSP%20is%20a%20list,newborn%20screening%20(NBS)%20programs
van Campen, Sollars, Thomas, Bartlett, Milano, & Parker et al. (2019). Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. International Journal Of Neonatal Screening, 5(4), 40. doi: 10.3390/ijns5040040
Adhikari, A. N., Gallagher, R. C., Wang, Y., Currier, R. J., Amatuni, G., Bassaganyas, L., Chen, F., Kundu, K., Kvale, M., Mooney, S. D., Nussbaum, R. L., Randi, S. S., Sanford, J., Shieh, J. T., Srinivasan, R., Sunderam, U., Tang, H., Vaka, D., Zou, Y., Koenig, B. A., … Brenner, S. E. (2020). The role of exome sequencing in newborn screening for inborn errors of metabolism. Nature medicine, 26(9), 1392–1397. https://doi.org/10.1038/s41591-020-0966-5
https://doi.org/10.1038/s41591-020-0966-5
Rajabi, F. (2018). Updates in Newborn Screening. Pediatric Annals, 47(5). doi: 10.3928/19382359-20180426-01
Wang, W., Yang, J., Xue, J., Mu, W., Zhang, X., Wu, W., Xu, M., Gong, Y., Liu, Y., Zhang, Y., Xie, X., Gu, W., Bai, J., & Cram, D. S. (2019). A comprehensive multiplex PCR based exome-sequencing assay for rapid bloodspot confirmation of inborn errors of metabolism. BMC medical genetics, 20(1), 3. https://doi.org/10.1186/s12881-018-0731-5
Waisbren, S., Bäck, D., Liu, C., Kalia, S., Ringer, S., Holm, I., & Green, R. (2014). Parents are interested in newborn genomic testing during the early postpartum period. Genetics In Medicine, 17(6), 501-504. doi: 10.1038/gim.2014.139
Pereira, S., Robinson, J. O., Gutierrez, A. M., Petersen, D. K., Hsu, R. L., Lee, C. H., Schwartz, T. S., Holm, I. A., Beggs, A. H., Green, R. C., McGuire, A. L., & BabySeq Project Group (2019). Perceived Benefits, Risks, and Utility of Newborn Genomic Sequencing in the BabySeq Project. Pediatrics, 143(Suppl 1), S6–S13. https://doi.org/10.1542/peds.2018-1099C
Pediatrics, 143(Suppl 1), S6–S13. https://doi.org/10.1542/peds.2018-1099C
Igenomix не имеет отношения к каким-либо новостям или публикациям, указанным выше. Освещение в новостях не подразумевает одобрение Igenomix.
Неонатальный скрининг новорожденных
Неонатальный скрининг проводят в родильном доме, однако некоторым детям по разным причинам его переносят на более поздние сроки, и тогда в амбулаторных условиях необходимо довести до родителей всю важность данного обследования и провести его.
Скрининг — быстрый, доступный, приблизительный метод обследования с целью выявления нуждающихся в более точной диагностике или помощи. В соответствии с этим общим понятием скрининг может иметь разные цели, в зависимости от которых определяется контингент обследуемых, охват, методы обследования. Скрининг в период новорождённости, который проводят в родильных домах (Приказ Минздравсоцразвития РФ от 22.03.2006 г. № 185 «О массовом обследовании новорождённых детей на наследственные заболевания». ), — это массовое обследование всех новорождённых с целью раннего выявления наследственных болезней для проведения их своевременного лечения.
), — это массовое обследование всех новорождённых с целью раннего выявления наследственных болезней для проведения их своевременного лечения.
Современный каталог болезней включает 2500 наследственных заболеваний. Среди них заметное место занимают наследственные болезни обмена веществ (800), которые, как правило, имеют тяжёлые и во многих случаях фатальные проявления, часто ведут к ранней инвалидизации детей. В то же время для многих наследственных болезней обмена веществ в настоящее время разработаны эффективные методы диагностики, а при ряде заболеваний — и лечения. В связи с этим массовое обследование детей в период новорождённости на наследственную патологию (неонатальный скрининг) имеет большое значение для раннего выявления этих болезней, своевременного лечения, профилактики инвалидности и развития тяжёлых клинических последствий, а также снижения летальности от наследственных заболеваний.
Фенилкетонурия (ФКУ) — наследственное заболевание, в основе которого лежит нарушение обмена аминокислоты фенилаланина, которая, накапливаясь в крови и спинномозговой жидкости, вызывает поражение нервной системы. Частота ФКУ среди новорождённых 1 на 5000-10 000 (в России — 1 на 6950). Отставание в развитии ребёнка выявляется во втором полугодии жизни. Примерно у 60% больных отмечают идиотию, у остальных — менее выраженные умственные нарушения. Раннее выявление заболеваний у новорождённых, своевременное и правильное лечение таких больных с первых дней жизни предупреждает задержку умственного развития детей. Диетотерапия — единственный эффективный метод лечения классической ФКУ. Она должна начинаться в течение первых трёх недель жизни ребёнка и продолжаться не менее 10 лет, если не всю жизнь.
Частота ФКУ среди новорождённых 1 на 5000-10 000 (в России — 1 на 6950). Отставание в развитии ребёнка выявляется во втором полугодии жизни. Примерно у 60% больных отмечают идиотию, у остальных — менее выраженные умственные нарушения. Раннее выявление заболеваний у новорождённых, своевременное и правильное лечение таких больных с первых дней жизни предупреждает задержку умственного развития детей. Диетотерапия — единственный эффективный метод лечения классической ФКУ. Она должна начинаться в течение первых трёх недель жизни ребёнка и продолжаться не менее 10 лет, если не всю жизнь.
Врождённый гипотиреоз — одно из наиболее часто встречающихся заболеваний щитовидной железы вследствие нарушения синтеза её гормонов. Его частота составляет 1 случай на 4000-5000 новорождённых. В России ежегодно рождается 400 детей с врожденным гипотиреозом. В основе заболевания лежит полная или частичная недостаточность тиреоидных гормонов, приводящая к задержке развития всех органов и систем. В первую очередь страдает ЦНС и интеллект. При поздней диагностике и несвоевременном лечении дети становятся инвалидами с полной утратой способности к обучению, трудоспособности и к социальной адаптации. Своевременно начатое лечение тиреоидными гормонами предотвращает развитие умственной отсталости. Эффективность лечения зависит от срока постановки диагноза, так как уже в первые месяцы жизни наступают необратимые изменения в умственном развитии и росте скелета.
При поздней диагностике и несвоевременном лечении дети становятся инвалидами с полной утратой способности к обучению, трудоспособности и к социальной адаптации. Своевременно начатое лечение тиреоидными гормонами предотвращает развитие умственной отсталости. Эффективность лечения зависит от срока постановки диагноза, так как уже в первые месяцы жизни наступают необратимые изменения в умственном развитии и росте скелета.
Адреногенитальный синдром — наследственное заболевание, обусловленное снижением активности фермента, участвующего в выработке гормонов надпочечника (кортизола и альдостерона). Распространённость, по данным разных авторов, колеблется от 1:5000 до 1:20 000 новорождённых. Клинические проявления зависят от того, на каком уровне блокируются ферменты. Наиболее тяжёлой, опасной для жизни является сольтеряющая форма, частота которой 1:27 000. Болезнь начинается в первую неделю жизни ребёнка, протекает остро, с выраженным обезвоживанием, падением артериального давления, судорогами и требует немедленного проведения реанимационных мероприятий с целью коррекции водно-электролитного баланса. При отсутствии адекватной терапии больные новорождённые умирают на 1-2-м месяце жизни. Для лечения назначают заместительную гормональную терапию (глюко- и минералкортикоиды).
При отсутствии адекватной терапии больные новорождённые умирают на 1-2-м месяце жизни. Для лечения назначают заместительную гормональную терапию (глюко- и минералкортикоиды).
Галактоземия — наследственное заболевание, связанное с невозможностью использования организмом углевода молока — галактозы. Частота болезни — 1 случай на 15 000-20 000 новорождённых. В основе её лежит отсутствие или резкое снижение активности ферментов, которые в процессе обмена веществ превращают галактозу молока в глюкозу. Вследствие неполного расщепления промежуточные продукты обмена оказывают токсическое воздействие на организм. Болезнь проявляется в виде тяжёлого поражения печени, нервной системы, глаз и других органов. Значительная часть больных, не получающих адекватной терапии, умирает в грудном возрасте; у других уже в первом полугодии жизни формируется тяжёлая инвалидизирующая патология: катаракта, цирроз печени, задержка нервнопсихического развития. При ранней постановке диагноза и своевременно начатом лечении сохраняется нормальный интеллект, не появляются нарушения глаз и печени. В настоящее время разработано и успешно применяется патогенетическое лечение диетой. Так как молоко (материнское и коровье) содержит лактозу, которая под действием ферментов расщепляется до галактозы и глюкозы, то с первых дней жизни, с момента установления диагноза, ребёнок должен быть переведён на безмолочное питание. Следует немедленно прекратить грудное вскармливание!
В настоящее время разработано и успешно применяется патогенетическое лечение диетой. Так как молоко (материнское и коровье) содержит лактозу, которая под действием ферментов расщепляется до галактозы и глюкозы, то с первых дней жизни, с момента установления диагноза, ребёнок должен быть переведён на безмолочное питание. Следует немедленно прекратить грудное вскармливание!
Часто задаваемые вопросы о скрининге капель крови новорожденных
Ответы на все ваши вопросы о скрининге капель крови новорожденных, в том числе о том, что происходит с карточкой капли крови вашего ребенка и почему иногда требуется второй образец.
Почему нужно брать кровь, когда детям всего около недели?
Каждому ребенку предлагается скрининг пятна крови новорожденного, также известный как тест на укол из пятки, в идеале в возрасте 5 дней. Новорожденных проверяют на: 9
Это лучшее время, чтобы проверить все состояния вместе. Это дает время, чтобы получить результаты, при необходимости провести диагностические тесты и начать любое необходимое лечение на ранней стадии.
Это дает время, чтобы получить результаты, при необходимости провести диагностические тесты и начать любое необходимое лечение на ранней стадии.
Если бы дети были протестированы позже, некоторые дети могли бы пострадать, чего можно было бы избежать, если бы они были протестированы раньше.
Младенцы, впервые приехавшие в страну или которым еще предстоит пройти тест на прокол пятки, могут пройти тест в любое время в возрасте до 1 года. Однако это не включает скрининг-тест на муковисцидоз, который не является надежным после 8-недельного возраста.
Новорожденные братья и сестры детей, страдающих одним из состояний, могут быть проверены на это конкретное состояние в возрасте до 5 дней. Затем на 5-й день можно взять еще один образец для выявления других состояний.
Узнайте больше о тесте капли крови новорожденных и о том, что включает в себя тест капли крови.
Когда следует проводить скрининг недоношенных детей?
У младенцев, поступивших в больницу до 5-го дня, возможно, из-за недоношенности или плохого самочувствия, при поступлении следует взять образец капли крови. Этот образец будет использоваться для скрининга серповидно-клеточной анемии в случае переливания крови ребенку.
Этот образец будет использоваться для скрининга серповидно-клеточной анемии в случае переливания крови ребенку.
У недоношенных или нездоровых детей, а также у детей, перенесших переливание крови, на 5-й день должен быть взят обычный образец капли крови. Этот образец крови записывается в карточке капли крови вместе с другой информацией.
Если ребенок родился до 32 недель, необходимо провести дополнительный тест на врожденный гипотиреоз в возрасте 28 дней или при выписке из больницы, в зависимости от того, что наступит раньше. Британский фонд щитовидной железы располагает дополнительной информацией о врожденном гипотиреозе.
Скрининг капли крови новорожденного
Кредит:
НАУЧНАЯ ФОТОГРАФИЯ / НАУЧНАЯ ФОТОБИБЛИОТЕКА https://www.sciencephoto.com/media/148998/view
Когда нужен второй образец?
Время от времени акушерка или патронажная сестра свяжутся с вами и попросят взять второй образец крови из пятки вашего ребенка.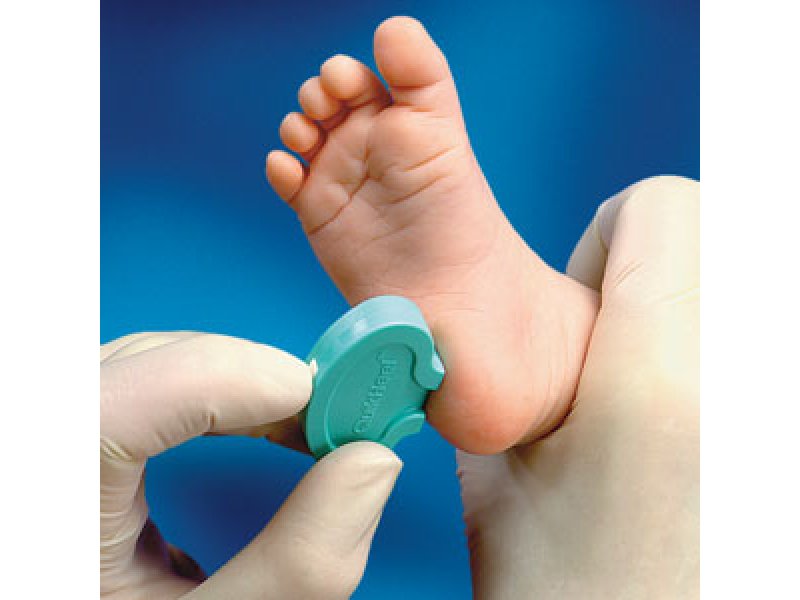 Это может быть связано с тем, что было собрано недостаточно крови, или результат был пограничным или неясным.
Это может быть связано с тем, что было собрано недостаточно крови, или результат был пограничным или неясным.
Это также может быть связано с тем, что ваш ребенок родился недоношенным. У детей, рожденных до 32 недель беременности, тестирование на 5-й день может не выявить врожденный гипотиреоз. Рекомендуется пройти еще один тест – либо в возрасте 28 дней, либо перед выпиской ребенка домой, в зависимости от того, что наступит раньше. Повторные результаты обычно нормальные.
Проверяются ли какие-либо состояния во время беременности?
Во время беременности вы пройдете обследование на серповидно-клеточную анемию. Можно проводить скрининг носителей муковисцидоза во время беременности, но в настоящее время это не делается в Великобритании.
Мне сказали , что мой ребенок является носителем. Что это значит для моего ребенка и моей семьи?
Статус носительства очень распространен и не означает, что ваш ребенок заболеет.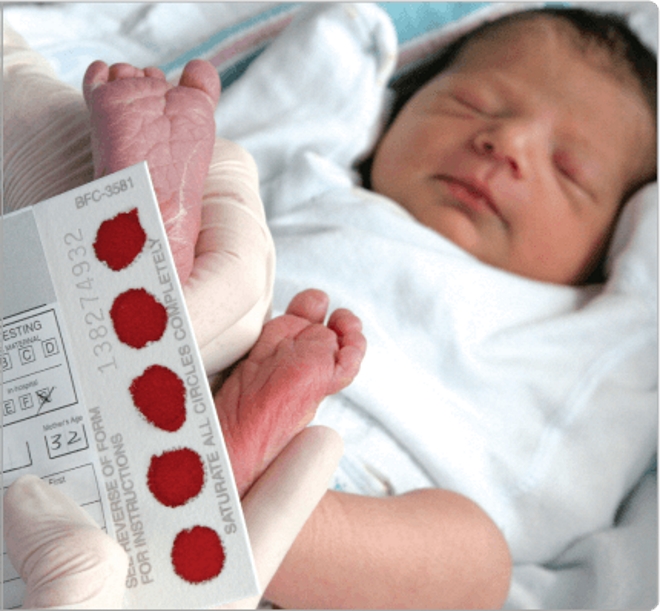
Для вашего ребенка:
- Они все еще должны быть здоровы. Иногда необходимы дополнительные тесты для подтверждения статуса носителя муковисцидоза. Вы можете организовать их через GP. Узнайте больше о том, как быть носителем муковисцидоза, на веб-сайте GOV.UK.
- Результаты исследования носителей серповидно-клеточной анемии обычно более очевидны. Ваша акушерка или врач общей практики сообщит вам, были ли рекомендованы дополнительные анализы, и они могут организовать их. Узнайте больше о том, когда ваш ребенок является носителем гена серповидно-клеточной анемии, на веб-сайте GOV.UK.
Для вашей семьи:
- Ребенок может быть носителем, потому что им является один из родителей. Это довольно распространенное явление и не должно влиять на их здоровье.
- В редких случаях ребенок может быть носителем, потому что оба его родителя унаследовали ген только от одного родителя. Если оба родителя являются носителями, существует вероятность 1 из 4, что будущие брат или сестра могут унаследовать 2 измененных гена и иметь наследственное заболевание.

- В настоящее время вам будет предложено пройти обследование на серповидно-клеточную анемию во время беременности, поэтому новые матери уже должны знать, являются ли они носителями. Консультант-генетик может объяснить это более подробно и организовать для вас анализы, если вы обеспокоены.
Если мы переедем в новый район, потребуются ли медицинским работникам доказательства того, что мой ребенок прошел скрининговый тест?
Если вы переезжаете в новый район, когда вашему ребенку меньше 1 года, вам необходимо будет предоставить врачу общей практики или патронажной сестре подтверждение того, что они прошли обследование. Это может быть письменная копия их результатов или результаты, записанные в личной медицинской карте ребенка.
Если вы переехали из-за границы и у вас нет доказательств прохождения тестирования, с вами можно обсудить и организовать повторное тестирование с вашего согласия.
Что такое карты крови новорожденных?
Эти карты используются не только для взятия образцов крови, но и для записи личной информации, включая имя ребенка, дату рождения, адрес и номер NHS; имя матери; и контактные данные врача общей практики и акушерки. Это необходимо, чтобы убедиться, что результаты скрининговых тестов соответствуют правильному ребенку.
Это необходимо, чтобы убедиться, что результаты скрининговых тестов соответствуют правильному ребенку.
Что происходит с карточкой крови моего ребенка?
После скрининга карты крови хранятся не менее 5 лет. Их можно использовать:
- для перепроверки результатов скрининга вашего ребенка
- для проведения других анализов, рекомендованных врачом
- для изучения генетических заболеваний, встречающихся в вашей семье
- для улучшения программы скрининга новорожденных
- для исследований чтобы помочь улучшить здоровье других младенцев и их семей в Великобритании
Это исследование не позволит идентифицировать вашего ребенка, а использование этих пятен крови регулируется сводом правил, который можно получить у вашей акушерки или посмотреть на на сайте GOV.UK.
Существует небольшая вероятность того, что исследователи захотят пригласить вас или вашего ребенка принять участие в исследовании, связанном с этой программой скрининга. Если вы не хотите, чтобы вас приглашали, сообщите об этом акушерке.
Если вы не хотите, чтобы вас приглашали, сообщите об этом акушерке.
Если у вас или вашего ребенка есть подозрения на серповидно-клеточную анемию или талассемию, информация будет передана в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS). Это помогает улучшить скрининг и профилактику или лечение заболевания.
Вы можете отказаться от регистрации или получить дополнительную информацию на сайтах gov.uk/phe/ncardrs и gov.uk/phe/newborn-outcomes.
Дополнительную информацию о том, зачем хранятся карточки для взятия проб крови и как защищается личная информация вашего ребенка, см. в карточках для взятия проб крови для новорожденных.
Заболевание мочи кленовым сиропом — NHS
Болезнь мочи кленового сиропа (MSUD) — редкое, но серьезное наследственное заболевание.
Это означает, что организм не может перерабатывать определенные аминокислоты («строительные блоки» белка), вызывая накопление вредных веществ в крови и моче.
Обычно наш организм расщепляет белковые продукты, такие как мясо и рыба, на аминокислоты. Любые аминокислоты, которые не нужны, обычно расщепляются и удаляются из организма.
Младенцы с MSUD не способны расщеплять аминокислоты, называемые лейцином, изолейцином и валином. Очень высокие уровни этих аминокислот вредны.
Одним из характерных симптомов MSUD является сладкий запах мочи, что и дало название этому состоянию.
В возрасте около 5 дней младенцам предлагается скрининг капли крови новорожденного, чтобы проверить наличие наследственных заболеваний, таких как MSUD. Это включает в себя прокол пятки вашего ребенка, чтобы собрать капли крови для анализа.
Если у вашего ребенка диагностирован MSUD, необходимо немедленно начать лечение, чтобы снизить риск серьезных осложнений. При ранней диагностике и правильном лечении можно значительно улучшить результат. Однако лечение MSUD необходимо продолжать пожизненно.
Однако лечение MSUD необходимо продолжать пожизненно.
Без лечения могут развиться тяжелые, опасные для жизни симптомы, включая судороги (припадки) или впадение в кому. Некоторые дети с нелеченым MSUD также подвержены риску повреждения головного мозга и задержки развития.
Симптомы MSUD обычно появляются в течение первых нескольких дней или недель после рождения. Более общие симптомы включают:
- сладкий запах мочи и пота
- плохой аппетит или потеря аппетита
- потеря веса
У детей с MSUD также могут быть эпизоды, известные как «метаболический кризис», иногда в начале их жизни. К симптомам метаболического кризиса относятся:
- упадок сил
- рвота
- раздражительность
- затрудненное дыхание
Важно немедленно обратиться за медицинской помощью, если у вашего ребенка появляются симптомы метаболического кризиса. Ваш врач даст вам совет, который поможет вам распознать признаки.
Ваш врач даст вам совет, который поможет вам распознать признаки.
У некоторых детей с MSUD могут не проявляться симптомы метаболического криза до конца первого года жизни или позже в детстве. Больница предоставит вам инструкции по неотложной помощи, которым нужно следовать, если ваш ребенок болен, что поможет предотвратить развитие этих симптомов.
Диета
Детей с диагнозом MSUD сначала направляют к специалисту-диетологу-метаболисту и назначают низкобелковую диету. Возможно, им тоже придется принимать лекарства. Диета разработана таким образом, чтобы уменьшить количество получаемых аминокислот, особенно лейцина, валина и изолейцина.
Продукты с высоким содержанием белка должны быть ограничены, в том числе:
- мясо
- рыба
- сыр
- яйца
- бобовые
- орехи
- вероятность развития заболевания 1 из 4; носитель MSUD
- шанс 1 из 4 получить пару нормальных генов
1 9020 этих продуктов для здорового роста и развития.
Некоторым детям необходимо принимать добавки изолейцина и валина наряду с предписанной диетой. Это помогает поддерживать здоровый уровень этих аминокислот в крови, не причиняя вреда. Для контроля этих уровней необходимы анализы крови.
Грудное молоко и детское питание также необходимо контролировать и измерять перед кормлением ребенка, как советует ваш диетолог. Обычная детская смесь содержит аминокислоты, количество которых необходимо ограничивать, поэтому вместо нее используется специальная смесь. Он содержит все витамины, минералы и другие аминокислоты, необходимые вашему ребенку.
Людям с MSUD необходимо всю оставшуюся жизнь соблюдать диету с низким содержанием белка, чтобы снизить риск метаболического кризиса. Когда ваш ребенок станет старше, ему в конечном итоге нужно будет научиться контролировать свой рацион, и он будет поддерживать связь с диетологом для получения рекомендаций и наблюдения.
Неотложная помощь
Если ваш ребенок заболеет, у него может случиться метаболический кризис. Это может привести к серьезному заболеванию и долгосрочному повреждению головного мозга, а также может быть опасным для жизни.
Можно снизить этот риск, перейдя на экстренную диету во время болезни.
Ваш диетолог предоставит подробные инструкции по низкобелковой диете и пищевым добавкам. Это может включать замену молока и продуктов, содержащих белок, специальными напитками с высоким содержанием сахара и прием добавок с аминокислотами.
Когда следует посетить больницу
Отвезите вашего ребенка в больницу, если развиваются симптомы метаболического кризиса, если он не может соблюдать экстренную диету и пищевые добавки, или если у него повторная диарея.
Свяжитесь с метаболической бригадой в больнице, чтобы сообщить им, что вы направляетесь прямо в отделение неотложной и неотложной помощи (A&E).
Возьмите любую информацию, которую вы получили о MSUD, в случае чрезвычайной ситуации на случай, если врачи раньше не посещали MSUD.
После того, как вы окажетесь в больнице, ваш ребенок может находиться под наблюдением и лечением с помощью жидкостей, вводимых непосредственно в вену (внутривенные жидкости).
Трансплантация печени
Трансплантация печени иногда является вариантом лечения MSUD. Если человек с MSUD получит донорскую печень, он больше не будет подвергаться риску метаболического кризиса и сможет нормально питаться.
Пересадка печени — серьезная процедура, сопряженная с определенными рисками.
Вам придется принимать лекарства для подавления иммунной системы (иммунодепрессанты) до конца жизни, чтобы организм не отторгал новую печень.
Важно взвесить все «за» и «против», прежде чем принимать решение о пересадке печени. Ваш врач сможет обсудить, является ли это подходящим вариантом.
Ваш врач сможет обсудить, является ли это подходящим вариантом.
Генетическое изменение (мутация), ответственное за MSUD, передается от родителей, у которых обычно нет никаких симптомов заболевания. Это называется аутосомно-рецессивным наследованием.
Это означает, что ребенку необходимо получить две копии измененных генов, чтобы у него развилось заболевание – одну от матери и одну от отца. Если ребенок получит только один мутировавший ген, он будет просто носителем MSUD.
Если вы являетесь носителем пораженных генов и у вас есть ребенок от партнера, который также является носителем, ваш ребенок имеет:
Хотя MSUD невозможно предотвратить, важно сообщить акушерке и врачу, если у вас есть семейный анамнез этого заболевания, чтобы как можно скорее были назначены анализы и назначено лечение.