
Скрининг из пятки новорожденного: Для чего нужен неонатальный скрининг?

Неонатальный скрининг в 2023 году: где и кому проводят и как берут анализ у новорожденного
Свежий номер
РГ-Неделя
Родина
Тематические приложения
Союз
Свежий номер
Общество
06.04.2023 17:21
Поделиться
Любовь Проценко
Правительство Москвы приняло решение расширить программу скрининга новорожденных. С этого года каждый ребенок проходит проверку на наличие у него 36 наследственных заболеваний или втрое большего количества нозологий, чем было до этого. Комментируя новшество, мэр Москвы Сергей Собянин написал в своем Telegram-канале: «Теперь врачи смогут начать лечение незамедлительно — это спасет многих малышей от развития патологий».
Денис Абрамов/РИА Новости
Напомним, в 2006 году неонатальный скрининг проводился по 5 нозологиям, в 2019 Москва включила в него уже 11, а теперь 36 нозологий.
Что позволило сделать такой заметный шаг вперед?
«Добавились заболевания, для борьбы с которыми у нас появились лекарства, лечебное питание, — рассказала корреспонденту «РГ» заведующая медико-генетическим отделением Морозовской детской городской клинической больницы Татьяна Кекеева. — Взять, например, спинально-мышечную атрофию. Ребенок, страдающий ею, не держит головку, не сидит, не встает. А причина в том, что у него в гене, ответственном за развитие нейронов, отсутствует участок. Но сейчас у нас есть так называемый геннозаместительный препарат, который оказывает колоссальный эффект, если начать применять его в первые полгода жизни. Есть и еще два препарата, так как и формы у этого заболевания бывают разные».
— Взять, например, спинально-мышечную атрофию. Ребенок, страдающий ею, не держит головку, не сидит, не встает. А причина в том, что у него в гене, ответственном за развитие нейронов, отсутствует участок. Но сейчас у нас есть так называемый геннозаместительный препарат, который оказывает колоссальный эффект, если начать применять его в первые полгода жизни. Есть и еще два препарата, так как и формы у этого заболевания бывают разные».
Именно в Морозовской детской больнице находится лаборатория, в которую и сдают на скрининг анализы новорожденных из всех роддомов Москвы — не только городского подчинения, но и федерального. А с этого года здесь проводится скрининг новорожденных еще и из Подмосковья, а также Магаданской области и Чукотского автономного округа — лаборатория получила статус межрегиональной.
Как проводят скрининг
Для анализа же берется кровь из пятки новорожденного и на специальных тест-полосках доставляется в Морозовскую детскую больницу. Но для этого нужно согласие родителей на скрининг.
Чем опасны генетические заболевания
Опасность генетических заболеваний заключается в том, что зачастую их симптомы появляются не сразу: может пройти месяц, полгода, а порой и год. И нередко по ним сразу и не поймешь, что происходит с малышом, доктору и маме с папой остается только гадать. Патология тем временем нарастает.
В Москве, например, за 2022 год обследовано 110 325 детей, у 99 из них были выявлены генетические заболевания. Семья узнала о проблемах со здоровьем малыша сразу: в случаях, когда ребенок не нуждается в срочной госпитализации, родителей приглашают в Морозовскую больницу на консультацию. Вместе с ними решается вопрос о дообследовании, если в нем есть необходимость, всех маленьких пациентов берут под наблюдение, их обеспечивают бесплатными лекарствами и лечебным питанием.
Российская газета — Столичный выпуск: №75(9020)
Поделиться
ЗдоровьеСемья и детиМосква
Newborn Screening — Russia
Неонатальный скрининг от Igenomix – это комплексный генетический тест, с помощью которого анализируются 237 генов методом NGS.

Заинтересовались?
Узнать больше
Наш email: [email protected]
Общая информация
- Неонатальный скрининг
- Преимущества
- Показания
Общая информация
- Около 3% -4% новорожденных имеют генетическое заболевание.
- Неонатальный скрининг – это пункт обязательной программы общественного здравоохранения, который обеспечивает исследования и последующее медицинское обслуживание при различных заболеваниях.
- Скрининговый тест Igenomix для новорожденных (IGX-NBS) – это комплексный генетический тест, с помщью которого анализируются 237 генов с использованием технологии секвенирования нового поколения (NGS), позволяющая напрямую определять генетические нарушения для быстрой и точной диагностики.
- Кроме того, этот тест определяет, является ли ребенок здоровым носителем любого из входящих в панель заболеваний.
Неонатальный скрининг от Igenomix предоставляет расширенную панель заболеваний, проанализированных с помощью технологий на основе NGS, предлагающих более широкий охват, чем подобные стандартные скрининги.
- Эти гены отвечают за нарушения развития, генетические и метаболические нарушения, которые вызывают серьезные проблемы со здоровьем, начиная с раннего детства.
- Главным преимуществом является раннее вмешательство с первого дня жизни для предотвращения умственной и физической инвалидизации, а также развития опасных для жизни заболеваний.
- Выявляется гораздо больше заболеваний, чем при обычном анализе крови.
Узнать больше
Цели обычного скрининга новорожденных
Цели скрининга новорожденных:
- Снижение заболеваемости и смертности от болезней, поддающихся лечению, путем проведения раннего вмешательства для улучшения неонатальных и долгосрочных результатов в отношении здоровья.
- Обеспечить всеобщее медицинское обследование всех новорожденных.
- Выявление новорожденных с положительным результатом скрининга.
- Диагностика заболеваний и состояний.
- Общение с семьями
- Направление в лечебные центры
- Долгосрочное наблюдение.

- Обучение врачей и пациентов.
Процесс проведения исследования?
Ограничения исследования
- Скрининг новорожденных НЕ заменяет генетическое тестированин на носительство моногенных заболеваний (CGT), а генетическое тестирование на носительство не заменяет скрининг новорожденных.
- Выполняется в первые дни жизни.
- Необходим образец крови (анализ слюны ожидает утверждения в Igenomix)
Критерии и классификация
Что было включено и почему?
Заболевания, включенные в скрининг новорожденных, были выбраны по следующим критериям:
- Заболевания с высокой распространенностью.
- Выявление болезней, при которых можно принять меры для улучшения прогноза.
- Отбор генов по данным доказательной медицины.
- Польза скрининга должна перевышать риски.
- Возможность скрининга для населения в целом на местном и национальном уровне.
- Наличие и доступ к лечению.
- Обучение врачей и повышение осведомленности общественности.

Скрининг новорожденных Igenomix по сравнению с текущими неонатальным скринингом
Наш неонатальный скрининг включает в себя все болезни из стандартного скрининга новорожденных (кровь из пятки новорожденного), а также из Рекомендованной унифицированной скрининговой панели (RUSP) и многое другое:
Заболевания, включенные в неонатальный скрининг от Igenomix, можно разделить на следующие группы. Вы можете найти пример болезни, включенной в каждую группу, и ее лечения.
Группа заболевания | Заболевание | Сиптомы | Лечение | Результат |
Заболевания обмена веществ | Синдром Менкеса | При отсутствии лечения характеризуется трудностями при кормлении, вялостью, рвотой, запахом кленового сиропа в ушной сере и моче, энцефалопатией и центральной дыхательной недостаточностью. | Терапия с помощью назначения диеты для снижения токсичных метаболитов и достижения необходимых концентраций недостающих аминокислот в плазме крови. | Бессимптомная нормальная жизнь |
Иммунодефициты | Тяжелый комбинированный иммунодефицит (ТКИД)
| Рецидивирующие, тяжело протекающие инфекции, вызванные условно-патогенными микроорганизмами, задержка развития | Защитные меры профилактики с помощью антибиотиков, противогрибковых, противовирусных препаратов и замены антител. | Снижение частоты инфекций, замедление прогрессирования заболевания и улучшение прогноза. |
Неврология | Болезнь Вильсона | Боли в животе, желтуха, гепатоспленомегалия, асцит, кровотечение из верхних отделов желудочно-кишечного тракта и поражение нервной системы. | Раннее начало хелатирования меди | Уменьшение накопления меди и предотвращение необратимых повреждений. |
Пульмонология | Муковисцидоз | Новорожденные страдают выпадением прямой кишки, мекониевой непроходимостью, респираторными инфекциями, недостаточностью поджелудочной железы, а также другими симптомами. | Раннее начало легочной терапии, диетотерапии, антибиотикопрофилактики, вакцинации, бронходилататоров. | Уменьшаетстя частота инфекций, поддерживается функция легких. Уменьшение прогрессирования заболевания и улучшение прогноза. |
Эндокринология | Врожденный гипотиреоз | У большинства младенцев болезнь протекает бессимптомно, у детей с симптомами обычно наблюдаются летаргия, хриплый крик, проблемы с приемом пищи, запоры, микседема, макроглоссия и другие симптомы. | Раннее начало заместительной терапии гормонов щитовидной железы (пероральный левотироксин) | Предотвращение развития нейрокогнитивных нарушений (измеряется коэффициеном IQ) |
Отоларингология | Наследственная тугоухость* | Новорожденные, скорее всего, не будут иметь симптомов до раннего детства, когда пациенту станет сложно понимать слова, слышать согласные и т. | Раннее начало использования слуховых аппаратов, логопеда и языковой терапии | Предотвращение задержки развития и речи |
Гемоглобинопатии | Серповидноклеточная анемия | Новорожденные могут иметь тяжелую анемию, вазоокклюзивный криз, хроническую боль в животе, гипоспленизм и инфекционные заболевания. | Раннее начало первичной профилактики острых осложнений, а также трансплантация гемопоэтических стволовых клеток | Предотвращение рецидивирующих острых вазоокклюзивных эпизодов и в сочетании с успешной трансплантацией гемопоэтических стволовых клеток добиться нормальной бессимптомной жизни. |
Нервно-мышечные заболевания | Спинальная мышечная атрофия | Характеризуется диффузной симметричной проксимальной мышечной слабостью, более выраженной в нижних конечностях, с отсутствием или заметно сниженными глубокими сухожильными рефлексами. | Раннее начало терапии, модифицирующей заболевание, и поддерживающей терапии. | Улучшение качества жизни и увеличение продолжительности жизни. |
 д.
д.
Все гены и заболевания
| ГЕН | ЗАБОЛЕВАНИЕ (OMIM) | ТИП ЗАБОЛЕВАНИЯ | ТИП НАСЛЕДОВАНИЯ |
| ABCD1 | Adrenoleukodystrophy | Metabolic disorder- Fatty acid oxidation | XL |
| ABCD4 | Methylmalonic aciduria and homocystinuria, cblJ type | Metabolic disorder- Organic acid | AR |
| ACAD8 | Isobutyryl-CoA dehydrogenase deficiency | Metabolic disorder- Organic acid | AR |
| ACADM | Acyl-CoA dehydrogenase, medium chain, deficiency of | Metabolic disorder- Fatty acid oxidation | AR |
| ACADS | Acyl-CoA dehydrogenase, short-chain, deficiency of | Metabolic disorder- Fatty acid oxidation | AR |
| ACADSB | 2-methylbutyrylglycinuria | Metabolic disorder- Organic acid | AR |
| ACADVL | VLCAD deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| ACAT1 | Alpha-methylacetoacetic aciduria | Metabolic disorder- Organic acid | AR |
| ADA | Severe combined immunodeficiency due to ADA deficiency | Immunodeficiency disorder | AR |
| ADK | Hypermethioninemia due to adenosine kinase deficiency | Metabolic disorder- Amino acid | AR |
| AHCY | Hypermethioninemia with deficiency of S-adenosylhomocysteine hydrolase | Metabolic disorder- Amino acid | AR |
| ALDh5A1 | Hyperprolinemia, type II | Metabolic disorder- Amino acid | AR |
| AMT | Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| ARG1 | Argininemia | Metabolic disorder- Amino acid | AR |
| ASL | Argininosuccinic aciduria | Metabolic disorder- Amino acid | AR |
| ASS1 | Citrullinemia | Metabolic disorder- Amino acid | AR |
| ATP7B | Wilson disease | Metabolic disorder- Copper | AR |
| AUH | 3-methylglutaconic aciduria, type I | Metabolic disorder- Organic acid | AR |
| BCKDHA | Maple syrup urine disease, type Ia | Metabolic disorder- Amino acid | AR |
| BCKDHB | Maple syrup urine disease, type Ib | Metabolic disorder- Amino acid | AR |
| BTD | Biotinidase deficiency | Metabolic disorder- Amino acid | AR |
| CBS | Homocystinuria, B6-responsive and nonresponsive types | Metabolic disorder- Amino acid | AR |
| CFTR | Cystic fibrosis | Respiratory disorder | AR |
| CPT1A | CPT deficiency, hepatic, type IA | Metabolic disorder- Fatty acid oxidation | AR |
| CPT2 | CPT II deficiency, lethal neonatal | Metabolic disorder- Fatty acid oxidation | AR |
| CTH | Cystathioninuria | Metabolic disorder- Amino acid | AR |
| CYP11B1 | Adrenal hyperplasia, congenital, due to 11-beta-hydroxylase deficiency | Endocrine disorder | AR |
| CYP21A2 | Adrenal hyperplasia, congenital, due to 21-hydroxylase deficiency | Endocrine disorder | AR |
| DBT | Maple syrup urine disease, type II | Metabolic disorder- Amino acid | AR |
| DNAJC12 | Hyperphenylalaninemia, mild, non-Bh5-deficient | Metabolic disorder- Amino acid | AR |
| DUOX2 | Thyroid dyshormonogenesis 6 | Endocrine disorder | AR |
| DUOXA2 | Thyroid dyshormonogenesis 5 | Endocrine disorder | AR |
| ETFA | Glutaric acidemia IIA | Metabolic disorder- Organic acid | AR |
| ETFB | Glutaric acidemia IIB | Metabolic disorder- Organic acid | AR |
| ETFDH | Glutaric acidemia IIC | Metabolic disorder- Organic acid | AR |
| FAH | Tyrosinemia, type I | Metabolic disorder- Amino acid | AR |
| GAA | Glycogen storage disease II | Metabolic disorder- Glicogen storage | AR |
| GALE | Galactose epimerase deficiency | Metabolic disorder- Carbohydrate | AR |
| GALK1 | Galactokinase deficiency with cataracts | Metabolic disorder- Carbohydrate | AR |
| GALT | Galactosemia | Metabolic disorder- Carbohydrate | AR |
| GCDH | Glutaric acidemia, type I | Metabolic disorder- Organic acid | AR |
| GCh2 | Hyperphenylalaninemia, Bh5-deficient, B | Metabolic disorder- Amino acid | AR |
| GCSH | ?Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| GJB2 | Deafness, digenic GJB2/GJB6 | Auditory disorder | AR |
| GJB6 | Deafness, digenic GJB2/GJB6 | Auditory disorder | AR |
| GLDC | Glycine encephalopathy | Metabolic disorder- Amino acid | AR |
| GNMT | Glycine N-methyltransferase deficiency | Metabolic disorder- Amino acid | AR |
| GSS | Glutathione synthetase deficiency | Metabolic disorder- Organic acid | AR |
| HADH | 3-hydroxyacyl-CoA dehydrogenase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HADHA | LCHAD deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HADHB | Trifunctional protein deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| HBB | Sickle cell anemia | Hemoglobin disorder | AR |
| HCFC1 | Mental retardation, X-linked 3 (methylmalonic acidemia and homocysteinemia, cblX type ) | Metabolic disorder- Organic acid | XLR |
| HGD | Alkaptonuria | Metabolic disorder- Amino acid | AR |
| HLCS | Holocarboxylase synthetase deficiency | Metabolic disorder- Organic acid | AR |
| HMGCL | HMG-CoA lyase deficiency | Metabolic disorder- Organic acid | AR |
| HPD | Tyrosinemia, type III | Metabolic disorder- Amino acid | AR |
| HSD17B10 | HSD10 mitochondrial disease | Metabolic disorder- Organic acid | XLD |
| IDUA | Mucopolysaccharidosis Ih/s | Metabolic disorder- Lysosomal enzime | AR |
| IL2RG | Severe combined immunodeficiency, X-linked | Immunodeficiency disorder | XLR |
| IVD | Isovaleric acidemia | Metabolic disorder- Organic acid | AR |
| LMBRD1 | Methylmalonic aciduria and homocystinuria, cblF type | Metabolic disorder- Organic acid | AR |
| MAT1A | Methionine adenosyltransferase deficiency, autosomal recessive | Metabolic disorder- Amino acid | AR |
| MCCC1 | 3-Methylcrotonyl-CoA carboxylase 1 deficiency | Metabolic disorder- Organic acid | AR |
| MCCC2 | 3-Methylcrotonyl-CoA carboxylase 2, deficiency | Metabolic disorder- Organic acid | AR |
| MCEE | Methylmalonyl-CoA epimerase deficiency | Metabolic disorder- Organic acid | AR |
| MLYCD | Malonyl-CoA decarboxylase deficiency | Metabolic disorder- Organic acid | AR |
| MMAA | Methylmalonic aciduria, vitamin B12-responsive | Metabolic disorder- Organic acid | AR |
| MMAB | Methylmalonic aciduria, vitamin B12-responsive, due to defect in synthesis of adenosylcobalamin, cblB complementation type | Metabolic disorder- Organic acid | AR |
| MMACHC | Methylmalonic aciduria and homocystinuria, cblC type | Metabolic disorder- Organic acid | AR |
| MMADHC | Methylmalonic aciduria and homocystinuria, cblD type | Metabolic disorder- Organic acid | AR |
| MMUT | Methylmalonic aciduria, mut(0) type | Metabolic disorder- Organic acid | AR |
| MTHFR | Homocystinuria due to MTHFR deficiency | Metabolic disorder- Amino acid | AR |
| MTR | Homocystinuria-megaloblastic anemia, cbl E type | Metabolic disorder- Amino acid | AR |
| MTRR | Homocystinuria due to MTHFR deficiency | Metabolic disorder- Amino acid | AR |
| MVK | Mevalonic aciduria | Metabolic disorder- Organic acid | AR |
| NADK2 | ?2,4-dienoyl-CoA reductase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| OTC | Ornithine transcarbamylase deficiency | Metabolic disorder- Amino acid | XLR |
| PAH | Phenylketonuria | Metabolic disorder- Amino acid | AR |
| PAX8 | Hypothyroidism, congenital, due to thyroid dysgenesis or hypoplasia | Endocrine disorder | AD |
| PCBD1 | Hyperphenylalaninemia, Bh5-deficient, D | Metabolic disorder- Amino acid | AR |
| PCCA | Propionic acidemia | Metabolic disorder- Organic acid | AR |
| PCCB | Propionic acidemia | Metabolic disorder- Organic acid | AR |
| PPM1K | ?Maple syrup urine disease, mild variant | Metabolic disorder- Organic acid | AR |
| PRDX1 | Methylmalonic aciduria and homocystinuria, cblC type, digenic | Metabolic disorder- Organic acid | AR |
| PRODH | Hyperprolinemia, type I | Metabolic disorder- Amino acid | AR |
| PTS | Hyperphenylalaninemia, Bh5-deficient, A | Metabolic disorder- Amino acid | AR |
| QDPR | Hyperphenylalaninemia, Bh5-deficient, C | Metabolic disorder- Amino acid | AR |
| RAG1 | Severe combined immunodeficiency, B cell-negative | Immunodeficiency disorder | AR |
| RAG2 | Severe combined immunodeficiency, B cell-negative | Immunodeficiency disorder | AR |
| SLC22A5 | Carnitine deficiency, systemic primary | Metabolic disorder- Fatty acid oxidation | AR |
| SLC25A13 | Citrullinemia, adult-onset type II | Metabolic disorder- Amino acid | AR |
| SLC25A20 | Carnitine-acylcarnitine translocase deficiency | Metabolic disorder- Fatty acid oxidation | AR |
| SLC3A1 | Cystinuria | Metabolic disorder- Amino acid | AR |
| SLC5A5 | Thyroid dyshormonogenesis 1 | Endocrine disorder | AR |
| SLC6A8 | Cerebral creatine deficiency syndrome 1 | Metabolic disorder- Fatty acid oxidation | XLR |
| SLC7A7 | Lysinuric protein intolerance | Metabolic disorder- Amino acid | AR |
| SLC7A9 | Cystinuria | Metabolic disorder- Amino acid | AR |
| SMN1 | Spinal muscular atrophy | Neuromuscular disorder | AR |
| TAT | Tyrosinemia, type II | Metabolic disorder- Amino acid | AR |
| TG | Thyroid dyshormonogenesis 3 | Endocrine disorder | AR |
| TPO | Thyroid dyshormonogenesis 2A | Endocrine disorder | AR |
| TSHB | Hypothyroidism, congenital, nongoitrous 4 | Endocrine disorder | AR |
| TSHR | Hypothyroidism, congenital, nongoitrous, 1 | Endocrine disorder | AR |
Ссылки на публикации
Связанные публикации:
ACOG Committee Opinion No. 778 Summary: Newborn Screening and the Role of the Obstetrician–Gynecologist. (2019). Obstetrics & Gynecology, 133(5), 1073-1074. doi: 10.1097/aog.0000000000003246
778 Summary: Newborn Screening and the Role of the Obstetrician–Gynecologist. (2019). Obstetrics & Gynecology, 133(5), 1073-1074. doi: 10.1097/aog.0000000000003246
https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html#:~:text=The%20RUSP%20is%20a%20list,newborn%20screening%20(NBS)%20programs
van Campen, Sollars, Thomas, Bartlett, Milano, & Parker et al. (2019). Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. International Journal Of Neonatal Screening, 5(4), 40. doi: 10.3390/ijns5040040
Adhikari, A. N., Gallagher, R. C., Wang, Y., Currier, R. J., Amatuni, G., Bassaganyas, L., Chen, F., Kundu, K., Kvale, M., Mooney, S. D., Nussbaum, R. L., Randi, S. S., Sanford, J., Shieh, J. T., Srinivasan, R., Sunderam, U., Tang, H., Vaka, D., Zou, Y., Koenig, B. A., … Brenner, S. E. (2020). The role of exome sequencing in newborn screening for inborn errors of metabolism. Nature medicine, 26(9), 1392–1397. https://doi.org/10.1038/s41591-020-0966-5
https://doi.org/10.1038/s41591-020-0966-5
Rajabi, F. (2018). Updates in Newborn Screening. Pediatric Annals, 47(5). doi: 10.3928/19382359-20180426-01
Wang, W., Yang, J., Xue, J., Mu, W., Zhang, X., Wu, W., Xu, M., Gong, Y., Liu, Y., Zhang, Y., Xie, X., Gu, W., Bai, J., & Cram, D. S. (2019). A comprehensive multiplex PCR based exome-sequencing assay for rapid bloodspot confirmation of inborn errors of metabolism. BMC medical genetics, 20(1), 3. https://doi.org/10.1186/s12881-018-0731-5
Waisbren, S., Bäck, D., Liu, C., Kalia, S., Ringer, S., Holm, I., & Green, R. (2014). Parents are interested in newborn genomic testing during the early postpartum period. Genetics In Medicine, 17(6), 501-504. doi: 10.1038/gim.2014.139
Pereira, S., Robinson, J. O., Gutierrez, A. M., Petersen, D. K., Hsu, R. L., Lee, C. H., Schwartz, T. S., Holm, I. A., Beggs, A. H., Green, R. C., McGuire, A. L., & BabySeq Project Group (2019). Perceived Benefits, Risks, and Utility of Newborn Genomic Sequencing in the BabySeq Project. Pediatrics, 143(Suppl 1), S6–S13. https://doi.org/10.1542/peds.2018-1099C
Pediatrics, 143(Suppl 1), S6–S13. https://doi.org/10.1542/peds.2018-1099C
Igenomix не имеет отношения к каким-либо новостям или публикациям, указанным выше. Освещение в новостях не подразумевает одобрение Igenomix.
Заболевание мочи кленовым сиропом — NHS
Болезнь мочи кленового сиропа (MSUD) — редкое, но серьезное наследственное заболевание.
Это означает, что организм не может перерабатывать определенные аминокислоты («строительные блоки» белка), вызывая накопление вредных веществ в крови и моче.
Обычно наш организм расщепляет белковые продукты, такие как мясо и рыба, на аминокислоты. Любые аминокислоты, которые не нужны, обычно расщепляются и удаляются из организма.
Младенцы с MSUD не могут расщеплять аминокислоты, называемые лейцином, изолейцином и валином. Очень высокие уровни этих аминокислот вредны.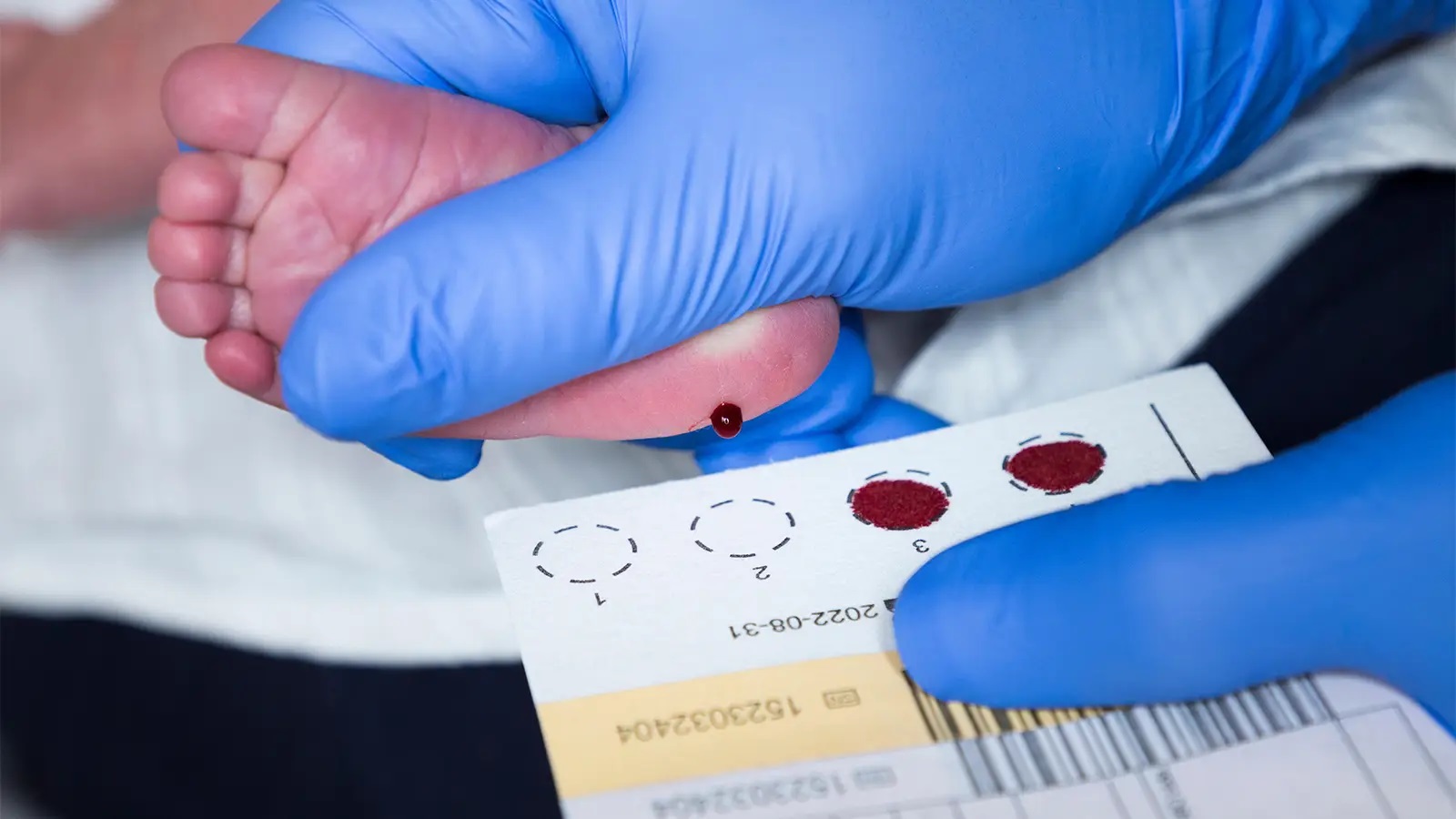
Одним из характерных симптомов MSUD является сладкий запах мочи, что и дало название этому состоянию.
В возрасте около 5 дней младенцам предлагается скрининг капли крови новорожденного, чтобы проверить наличие наследственных заболеваний, таких как MSUD. Это включает в себя прокол пятки вашего ребенка, чтобы собрать капли крови для анализа.
Если у вашего ребенка диагностирован MSUD, необходимо немедленно начать лечение, чтобы снизить риск серьезных осложнений. При ранней диагностике и правильном лечении можно значительно улучшить результат. Однако лечение MSUD необходимо продолжать пожизненно.
Без лечения могут развиться тяжелые, опасные для жизни симптомы, включая судороги (припадки) или впадение в кому. Некоторые дети с нелеченым MSUD также подвержены риску повреждения головного мозга и задержки развития.
Симптомы MSUD обычно появляются в течение первых нескольких дней или недель после рождения. К более общим симптомам относятся:
К более общим симптомам относятся:
- сладкий запах мочи и пота
- плохой аппетит или потеря аппетита
- потеря веса
У детей с MSUD также могут быть эпизоды, известные как «метаболический кризис», иногда в начале их жизни. К симптомам метаболического кризиса относятся:
- упадок сил
- рвота
- раздражительность
- затрудненное дыхание
Важно немедленно обратиться за медицинской помощью, если у вашего ребенка появляются симптомы метаболического кризиса. Ваш врач даст вам совет, который поможет вам распознать признаки.
У некоторых детей с MSUD могут не проявляться симптомы метаболического кризиса до конца первого года жизни или позже в детстве. Больница предоставит вам инструкции по неотложной помощи, которым нужно следовать, если ваш ребенок болен, что поможет предотвратить развитие этих симптомов.
Диета
Детей с диагнозом MSUD сначала направляют к специалисту-диетологу-метаболисту и назначают низкобелковую диету. Возможно, им тоже придется принимать лекарства. Диета разработана таким образом, чтобы уменьшить количество получаемых аминокислот, особенно лейцина, валина и изолейцина.
Необходимо ограничить продукты с высоким содержанием белка, в том числе:
- мясо
- рыба
- сыр
- яйца
- бобовые
- орехи
Ваш врач-диетолог предоставит подробные рекомендации и рекомендации, так как вашему ребенку все еще нужно немного этих продуктов для здорового роста и развития.
Некоторым детям необходимо принимать добавки изолейцина и валина наряду с предписанной диетой. Это помогает поддерживать здоровый уровень этих аминокислот в крови, не причиняя вреда. Для контроля этих уровней необходимы анализы крови.
Это помогает поддерживать здоровый уровень этих аминокислот в крови, не причиняя вреда. Для контроля этих уровней необходимы анализы крови.
Грудное молоко и детская смесь также должны контролироваться и измеряться перед кормлением ребенка, как советует ваш диетолог. Обычная детская смесь содержит аминокислоты, количество которых необходимо ограничивать, поэтому вместо нее используется специальная смесь. Он содержит все витамины, минералы и другие аминокислоты, необходимые вашему ребенку.
Людям с MSUD необходимо всю оставшуюся жизнь соблюдать диету с низким содержанием белка, чтобы снизить риск метаболического кризиса. Когда ваш ребенок станет старше, ему в конечном итоге нужно будет научиться контролировать свой рацион, и он будет поддерживать связь с диетологом для получения рекомендаций и наблюдения.
Неотложная помощь
Если ваш ребенок заболеет, у него может случиться метаболический кризис. Это может привести к серьезному заболеванию и долгосрочному повреждению головного мозга, а также может быть опасным для жизни.
Это может привести к серьезному заболеванию и долгосрочному повреждению головного мозга, а также может быть опасным для жизни.
Можно снизить этот риск, перейдя на экстренную диету во время болезни.
Ваш диетолог предоставит подробные инструкции по низкобелковой диете и пищевым добавкам. Это может включать замену молока и продуктов, содержащих белок, специальными напитками с высоким содержанием сахара и прием добавок с аминокислотами.
Когда следует посетить больницу
Отвезите вашего ребенка в больницу, если у него развиваются симптомы метаболического кризиса, если он не может соблюдать экстренную диету и пищевые добавки, или если у него повторная диарея.
Свяжитесь с метаболической командой в больнице, чтобы сообщить им, что вы направляетесь прямо в отделение неотложной и неотложной помощи (A&E).
Возьмите любую информацию, которую вы получили о MSUD, в случае возникновения чрезвычайной ситуации, если врачи ранее не посещали MSUD.
После того, как вы окажетесь в больнице, ваш ребенок может находиться под наблюдением и лечиться с помощью жидкостей, вводимых непосредственно в вену (внутривенные жидкости).
Трансплантация печени
Трансплантация печени иногда является вариантом лечения MSUD. Если человек с MSUD получит донорскую печень, он больше не будет подвергаться риску метаболического кризиса и сможет нормально питаться.
Пересадка печени — серьезная процедура, сопряженная с определенными рисками.
Вам придется принимать лекарства для подавления иммунной системы (иммунодепрессанты) до конца жизни, чтобы организм не отторгал новую печень.
Важно взвесить все «за» и «против», прежде чем принимать решение о пересадке печени. Ваш врач сможет обсудить, является ли это подходящим вариантом.
Генетическое изменение (мутация), ответственное за MSUD, передается от родителей, у которых обычно нет никаких симптомов заболевания. Это называется аутосомно-рецессивным наследованием.
Это называется аутосомно-рецессивным наследованием.
Это означает, что ребенку необходимо получить две копии измененных генов, чтобы у него развилось заболевание: одну от матери и одну от отца. Если ребенок получит только один мутировавший ген, он будет просто носителем MSUD.
Если вы являетесь носителем пораженных генов и у вас есть ребенок от партнера, который также является носителем, ваш ребенок имеет:
- вероятность развития заболевания 1 из 4; носитель MSUD
- шанс 1 из 4 получить пару нормальных генов
Несмотря на то, что MSUD невозможно предотвратить, важно сообщить акушерке и врачу, если у вас есть семейный анамнез заболевания, чтобы как можно скорее были назначены анализы и назначено лечение.
Вы также можете рассмотреть генетическое и геномное тестирование. Это может привести к направлению на генетическое консультирование, которое предлагает поддержку, информацию и советы о генетических заболеваниях.
Если у вашего ребенка MSUD, ваша медицинская бригада передаст информацию о нем или ней в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать лучшие способы профилактики и лечения этого заболевания. Вы можете отказаться от регистрации в любое время.
GOV.UK: брошюра для пациентов Национальной службы регистрации врожденных аномалий и редких заболеваний.
Последняя проверка страницы: 20 сентября 2021 г.
Дата следующей проверки: 20 сентября 2024 г.
Информация о программе скрининга новорожденных: О скрининге новорожденных
На этой странице:
Скрининг новорожденных в Миннесоте
Как работает скрининг новорожденных
Скрининг новорожденных в штате Миннесота
С 1964 года Департамент здравоохранения Миннесоты (MDH) проводит скрининг новорожденных штата Миннесота вскоре после рождения, чтобы определить, подвержены ли они риску развития редких скрытых заболеваний. Если не лечить, эти расстройства могут привести к болезни, инвалидности, задержке развития или смерти. Однако при раннем выявлении этих нарушений вмешательства, прием лекарств или изменение диеты могут помочь предотвратить большинство проблем со здоровьем, вызванных нарушениями, выявленными в группе скрининга новорожденных.
Если не лечить, эти расстройства могут привести к болезни, инвалидности, задержке развития или смерти. Однако при раннем выявлении этих нарушений вмешательства, прием лекарств или изменение диеты могут помочь предотвратить большинство проблем со здоровьем, вызванных нарушениями, выявленными в группе скрининга новорожденных.
Миннесота — национальный лидер в области скрининга новорожденных. Программа скрининга новорожденных совместно с больницами, лабораториями и медицинскими работниками по всему штату проводит скрининг новорожденных на наличие более 60 нарушений, влияющих на обмен веществ, гормоны, иммунную систему, кровь, дыхание, пищеварение, слух или сердце.
Наши цели:
- Обследовать всех младенцев штата Миннесота.
- Для выявления младенцев с редкими скрытыми нарушениями или с потерей слуха в раннем возрасте, когда лечение и вмешательство могут предотвратить проблемы со здоровьем, способствовать развитию и спасти жизни.
- Обеспечить, чтобы все младенцы с аномальными результатами скрининга новорожденных имели доступ к быстрой диагностической оценке и услугам раннего вмешательства.
- Усовершенствовать системы последующего наблюдения, отчетности и подключения к службам для младенцев, выявленных при скрининге новорожденных.
Как работает скрининг новорожденных
Скрининг новорожденных в Миннесоте состоит из трех простых тестов: скрининг капли крови, скрининг слуха и скрининг пульсоксиметрии. В этом коротком видео из программы «Первый тест ребенка» объясняется, чего ожидать в процессе скрининга новорожденных. См. приведенную ниже информацию для получения более подробной информации о каждом экране.
Что ожидать от первого теста вашего ребенка Видео Текст
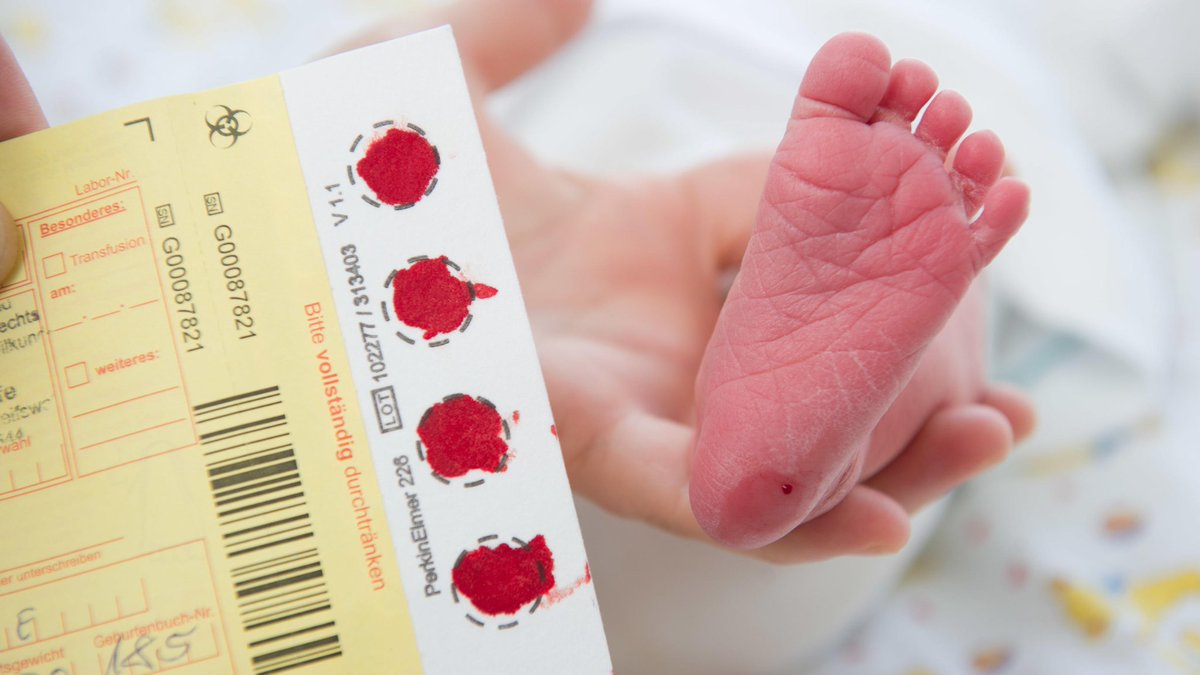
Скрининг пятна крови
Когда новорожденному будет от 24 до 48 часов, медицинский работник возьмет несколько капель крови из пятки новорожденного. Многие люди называют этот процесс «пяточной палочкой». Капли крови заполняют пять точек на карточке из фильтровальной бумаги. После высыхания капель крови их отправляют в нашу программу Министерства здравоохранения Миннесоты для проверки на более 50 наследственных и врожденных заболеваний.Результаты скрининга капли крови отправляются по почте в родильное учреждение или вызываются у поставщика первичной медико-санитарной помощи новорожденного, если необходимо дальнейшее тестирование.
Многие люди называют этот процесс «пяточной палочкой». Капли крови заполняют пять точек на карточке из фильтровальной бумаги. После высыхания капель крови их отправляют в нашу программу Министерства здравоохранения Миннесоты для проверки на более 50 наследственных и врожденных заболеваний.Результаты скрининга капли крови отправляются по почте в родильное учреждение или вызываются у поставщика первичной медико-санитарной помощи новорожденного, если необходимо дальнейшее тестирование.
Скрининг слуха
Пока новорожденный спит, медицинский работник проведет простой тест для проверки потери слуха в диапазоне, где слышна речь. Небольшое скрининговое устройство будет воспроизводить тихие звуки и измерять реакцию новорожденного. Скрининг лучше всего проводить как можно скорее, через 12 часов после родов, и его следует завершить до того, как новорожденному исполнится один месяц.
Скрининг пульсоксиметрии
Когда новорожденному исполнится хотя бы один день, медицинский работник проведет быстрый и простой тест, называемый пульсоксиметрией.