
Синдром ушера: CDH23, CLRN1, DFNB31 (WHRN), GPR98, MYO7A, PCDH15, USH1C, USH1G, USH2A

Not Found (#404)
Not Found (#404)
|
Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов | Иванова М.
 Е., Атарщиков Д.С., Демчинский А.М., Стрельников В.В., Бар Д., Порядин Г.В., Балашова Л.М., Салмаси Ж.М.
Е., Атарщиков Д.С., Демчинский А.М., Стрельников В.В., Бар Д., Порядин Г.В., Балашова Л.М., Салмаси Ж.М.
В статье представлены результаты исследования, посвященного изучению спектра мутаций российской когорты пациентов с синдромом Ашера и проведению анализа метаболома и интерактома, а также поиска путей таргетного лечения синдрома Ашера
Введение
Синдром Ашера (Usher syndrome, USH) является аутосомно-рецессивным заболеванием, на которое приходится почти половина всех случаев сочетанной наследственной глухоты и слепоты. По оценкам, распространенность USH составляет от 3,2 до 6,2 на 100 000 [1]. Тем не менее распространенность может быть до 1 на 6000. USH проявляется нарушением слуха, вестибулярной дисфункцией и пигментным ретинитом (RP) (синоним — тапеторетинальная абиотрофия сетчатки). В зависимости от возраста начала и тяжести клинических проявлений USH подразделяют на три основных клинических подтипа: USh2, USh3 и USh4. USh2 является наиболее тяжелой формой, наблюдающейся с рождения или с манифестацией до 5 лет, которая возникает в результате мутаций в любом из 6 генов: MYO7A, USh2C, CDh33, PCDh25, USh2G и CIB2. USh3 связан с мутациями в 3 генах: USh3A, ADGRV1 и DFNB31. USh4 вызван мутациями в гене CLRN1. Недавно был выделен 4-й атипичный USH, который возникает в результате мутаций в любом из генов: HARS, CEP250, PDZD7, C2orf71. На долю USh2 приходится около трети всех зарегистрированных случаев USH, на долю USh3 — две трети. USh4 протекает клинически наиболее благоприятно, встречается очень редко (~3%).
USh3 связан с мутациями в 3 генах: USh3A, ADGRV1 и DFNB31. USh4 вызван мутациями в гене CLRN1. Недавно был выделен 4-й атипичный USH, который возникает в результате мутаций в любом из генов: HARS, CEP250, PDZD7, C2orf71. На долю USh2 приходится около трети всех зарегистрированных случаев USH, на долю USh3 — две трети. USh4 протекает клинически наиболее благоприятно, встречается очень редко (~3%).
В течение последних нескольких лет были предприняты значительные усилия для описаний молекулярных характеристик, спектра мутаций и диагностики USH в различных популяциях, в т. ч. в испанской [2], американской [3], европейской [1], датской [4], финской [5], британской [1], алжирской [6], итальянской [7], немецкой [8], арабской [9], африканской [6] и китайской [10]. Современные методы секвенирования следующего поколения (next generation sequencing — NGS) значительно улучшили эффективность молекулярной характеристики в USH [1, 3, 11–13]. Кроме того, традиционный метод Сэнгера и метод мультиплексной амплификации (Multiplex ligation-dependent probe amplification — MLPA) успешно применяются для выявления глубоких интронных мутаций, больших делеций/дупликаций и вариаций числа копий генов (copy number variation — CNV).
Кроме того, традиционный метод Сэнгера и метод мультиплексной амплификации (Multiplex ligation-dependent probe amplification — MLPA) успешно применяются для выявления глубоких интронных мутаций, больших делеций/дупликаций и вариаций числа копий генов (copy number variation — CNV).
Работы, в которых были представлены молекулярный профиль пациентов при центральных дистрофиях сетчатки в российской популяции [14] и генотип/фенотип корреляции в единичных клинических случаях пациентов с USH [15], уже были опубликованы авторами. Тем не менее нет опубликованных данных по USH в российской когорте в соответствии с утвержденным протоколом клинических исследований.
Цель исследования: изучить спектр мутаций российской когорты пациентов с USH в рамках наблюдательного исследования NCT03319524 [16] и провести анализ метаболома и интерактома, а также патогенетических путей развития USH для поиска путей таргетного лечения.
Материал и методы
28 пациентов из группы 3214 пациентов (11 мужчин, 17 женщин; возраст — 47,6±12,7 года; возрастной диапазон проявления заболевания — 0–18 лет) с наиболее выраженными клиническими симптомами USH (нейросенсорная тугоухость 3–4 степени и сужение полей зрения до 5–15 градусов) были приглашены к участию. Национальный состав группы: 21 пациент (75%) — русские, 3 (10%) — украинцы, 2 (7%) — евреи, 1 (4%) — белорус и 1 (4%) — чуваш. Письменное информированное согласие было получено у всех 28 пациентов, исследование было одобрено Независимым международным этическим комитетом (Москва).
Национальный состав группы: 21 пациент (75%) — русские, 3 (10%) — украинцы, 2 (7%) — евреи, 1 (4%) — белорус и 1 (4%) — чуваш. Письменное информированное согласие было получено у всех 28 пациентов, исследование было одобрено Независимым международным этическим комитетом (Москва).
Клиническое обследование: каждому пациенту проводилась биомикроскопия, периметрия, визометрия, оптическая когерентная томография (ОКТ), офтальмоскопия с фоторегистрацией, электроретинография (ЭРГ), запись зрительно вызванных потенциалов, 32-оттеночный цветотест Хью, тональная и электронная аудиометрия, измерение акустического импеданса, вестибулометрия, электронистагмография и постурометрия. Молекулярно-генетическое подтверждение клинического диагноза выполнено методом высокопроизводительного параллельного секвенирования ДНК. Был проведен подробный компьютерный анализ патогенетических путей развития клинической картины у 20 пациентов с подтвержденным диагнозом USH. Использовалось программное обеспечение HGVS, Genemap, Genemania, CLINVAR, Genego, STRING, Cytoscape и база данных консорциума EMBL для моделирования интерактома и анализа полученных данных.
Результаты и обсуждение
Частота обнаруженных мутаций в обследованной когорте пациентов составила: 50% в гене USh3A, 39% — MYO7A, 5% — PCDh25, 3% — USh2C, 3% — CDh33, характеристика которых представлена авторами ранее [17] (табл. 1). Для обсуждения терапевтических подходов к коррекции утраченных или недостаточных функций генов в данной группе пациентов необходимо изучение и сопоставление всех факторов, которые влияют на патогенез заболевания, в т. ч. учет молекулярно-генетических факторов и факторов внешней среды: (геном —> транскриптом —> протеом —> метаболом), в совокупности образующих интерактом, которые определяют фенотип того или иного пациента [18]. Регуляции метаболизма в сетчатке обеспечиваются:
1) количеством ферментов в зависимости от активации или ингибирования транскрипции и синтеза белков; 2) каталитической активностью ферментов, что контролируется аллостерическими модуляторами, активаторами, конкурентными ингибиторами (т. е. агонистами и антагонистами) и посттрансляционными модификациями, такими как фосфорилирование, ацетилирование и гликозилирование под контролем гормонов, ростовых факторов и нейротрансмиттеров; 3) субстратной доступностью (концентрацией), которая зависит от систем активного мембранного транспорта (помпы, транспортеры, требующие затрат энергии) и пассивной диффузии через субстрат-специфические мембранные белки (ионные каналы), которые контролируются концентрационным градиентом. В процессе фототрансдукции происходит передача зрительного сигнала, начиная с захвата фотонов сетчаткой глаза и завершая формированием зрительных образов в зрительной коре головного мозга. Подробнее о геномном контроле фототрансдукции можно узнать, изучая функцию каждого из белков фототрансдукции, транслируемых с указанных генов, сначала по отдельности, а затем собирая в одну общую сеть интерактома. Изучение интерактома, т. е. полного набора взаимодействий между молекулами в отдельной клетке, как непосредственных физических контактов между белками (белок-белковые взаимодействия), так и непрямых взаимодействий генов (например, эпистаз, ап-регулирование, ко-активация, влияние факторов транскрипции и пр.
е. агонистами и антагонистами) и посттрансляционными модификациями, такими как фосфорилирование, ацетилирование и гликозилирование под контролем гормонов, ростовых факторов и нейротрансмиттеров; 3) субстратной доступностью (концентрацией), которая зависит от систем активного мембранного транспорта (помпы, транспортеры, требующие затрат энергии) и пассивной диффузии через субстрат-специфические мембранные белки (ионные каналы), которые контролируются концентрационным градиентом. В процессе фототрансдукции происходит передача зрительного сигнала, начиная с захвата фотонов сетчаткой глаза и завершая формированием зрительных образов в зрительной коре головного мозга. Подробнее о геномном контроле фототрансдукции можно узнать, изучая функцию каждого из белков фототрансдукции, транслируемых с указанных генов, сначала по отдельности, а затем собирая в одну общую сеть интерактома. Изучение интерактома, т. е. полного набора взаимодействий между молекулами в отдельной клетке, как непосредственных физических контактов между белками (белок-белковые взаимодействия), так и непрямых взаимодействий генов (например, эпистаз, ап-регулирование, ко-активация, влияние факторов транскрипции и пр. ), позволяет выделять наиболее перспективные терапевтические мишени. В то время как данные об экспрессии матричной рибонуклеиновой кислоты (мРНК) генов и данные протеомного анализа не раскрывают полностью всего того, что может происходить в клетке, метаболические профили (изучение метаболома сетчатки) [19, 20] могут дать мгновенный снимок физиологических процессов в клетке. Одна из задач современной медицины, системной биологии и функциональной геномики — это интегрирование данных протеомики, транскриптомики и метаболической информации для получения более целостного представления о строении и функции сетчатки у конкретного пациента. При компьютерном анализе и изучении взаимодействия наиболее активных генов метаболизма сетчатки при USH в изучаемой когорте выяснилось, что в 67% случаев наблюдается физическое взаимодействие генов, в 13% — ко-экспрессия, в 6% — ко-локализация, в 4% — совместные метаболические пути, в 1,4% — генные взаимодействия, 0,59% имеют общие белковые домены. В 6% случаев взаимодействие генов или белков, синтезируемых с этих генов, не доказано, но по некоторым предсказательным алгоритмам их взаимодействие вероятно.
), позволяет выделять наиболее перспективные терапевтические мишени. В то время как данные об экспрессии матричной рибонуклеиновой кислоты (мРНК) генов и данные протеомного анализа не раскрывают полностью всего того, что может происходить в клетке, метаболические профили (изучение метаболома сетчатки) [19, 20] могут дать мгновенный снимок физиологических процессов в клетке. Одна из задач современной медицины, системной биологии и функциональной геномики — это интегрирование данных протеомики, транскриптомики и метаболической информации для получения более целостного представления о строении и функции сетчатки у конкретного пациента. При компьютерном анализе и изучении взаимодействия наиболее активных генов метаболизма сетчатки при USH в изучаемой когорте выяснилось, что в 67% случаев наблюдается физическое взаимодействие генов, в 13% — ко-экспрессия, в 6% — ко-локализация, в 4% — совместные метаболические пути, в 1,4% — генные взаимодействия, 0,59% имеют общие белковые домены. В 6% случаев взаимодействие генов или белков, синтезируемых с этих генов, не доказано, но по некоторым предсказательным алгоритмам их взаимодействие вероятно.
Анализ интерактома (рис. 1) показывает, что у протокадгерина (PCDh25) большая сеть контактов с кадгеринами (CDh33), у ушерина (USh3A) — с разными видами коллагенов (COL4A3), у миозина (MYO7A) — с вирлиновым комплексом (WHRN) и различными подвидами актина (ACTA2, ACTG2). Примечательно, что MYO7A взаимодействует с гармонином, что позволяет предположить, что он может участвовать в закреплении верхнего конца цилии стержня фоторецептора (видоизмененные реснички). MYO7A также взаимодействует с некоторыми сплайс-формами протокадгерина (PCDh25), предполагается, что этот моторный белок может участвовать в формировании комплексов на обоих концах реснички (рис. 2).
MYO7A — ген, кодирует одну из форм белка миозина, расположен на 11-й хромосоме и имеет 55 экзонов. Миозин действует как моторный белок, управляемый АТФазой, для транспорта меланосом и фагосом вдоль нитей актина в пигментном эпителии сетчатки, а также для транспорта опсина и других белков фототрансдукции в фоторецепторах. Мутации в гене MYO7A вызывают тяжелую и глубокую непрогрессирующую нейросенсорную тугоухость, пигментный ретинит до начала подросткового периода и часто с вестибулярной арефлексией. Острота центрального зрения обычно лучше, чем 0,3, в первые два десятилетия жизни. Поля зрения у пациентов с USh2B начинают сужаться в средней периферии и переходят к остаточным небольшим центральным островкам с небольшими участками периферических полей зрения на самых поздних стадиях заболевания. Офтальмоскопически у пациентов наблюдаются стушеванность артерий и атрофия ретинального пигментного эпителия (РПЭ) с пигментацией по типу «костных телец». Аутофлуоресценция глазного дна может показать кольцо гипераутофлуоресценции в макуле. ОКТ часто выявляет потерю наружных слоев сетчатки и присутствие кистозного макулярного отека. Ганцфельд-ЭРГ почти всегда нерегистрируемая или амплитуды сигналов сильно снижены.
Мутации в гене MYO7A вызывают тяжелую и глубокую непрогрессирующую нейросенсорную тугоухость, пигментный ретинит до начала подросткового периода и часто с вестибулярной арефлексией. Острота центрального зрения обычно лучше, чем 0,3, в первые два десятилетия жизни. Поля зрения у пациентов с USh2B начинают сужаться в средней периферии и переходят к остаточным небольшим центральным островкам с небольшими участками периферических полей зрения на самых поздних стадиях заболевания. Офтальмоскопически у пациентов наблюдаются стушеванность артерий и атрофия ретинального пигментного эпителия (РПЭ) с пигментацией по типу «костных телец». Аутофлуоресценция глазного дна может показать кольцо гипераутофлуоресценции в макуле. ОКТ часто выявляет потерю наружных слоев сетчатки и присутствие кистозного макулярного отека. Ганцфельд-ЭРГ почти всегда нерегистрируемая или амплитуды сигналов сильно снижены.
В сетчатке большая часть молекул миозина MYO7A находится в пигментном эпителии сетчатки (РПЭ), где происходят многие реакции зрительного ретиноидного цикла. V.S. Lopes et al. наблюдали [21], что сетчатка с мутациями в MYO7A устойчива к острому световому повреждению по причине более низкого уровня RPE65, изомеразы RPE, которая играет ключевую роль в ретиноидном цикле. Было показано, что RPE65 обычно подвергается светозависимой транслокации, чтобы стать более сконцентрированным в центральной области клеток RPE. Эта транслокация требует участия моторного белка MYO7A, поэтому при мутациях в гене MYO7A RPE65 частично локализован на свету и распадается быстрее, возможно, из-за его неправильной локализации, что дает правдоподобное объяснение его более низким уровням. После 50–60% фотозасвета сетчатка с мутацией в MYO7A демонстрирует повышенные уровни всех трансретиниловых эфиров на начальных стадиях темнового восстановления, что согласуется с дефицитом активности RPE65. Наконец, MYO7A и RPE65 совместно иммунопреципитируются из клеточного лизата RPE антителами против любого из белков, и два белка были локализованы в одних и тех же участках клетки, что указывает на их прямое или косвенное взаимодействие.
V.S. Lopes et al. наблюдали [21], что сетчатка с мутациями в MYO7A устойчива к острому световому повреждению по причине более низкого уровня RPE65, изомеразы RPE, которая играет ключевую роль в ретиноидном цикле. Было показано, что RPE65 обычно подвергается светозависимой транслокации, чтобы стать более сконцентрированным в центральной области клеток RPE. Эта транслокация требует участия моторного белка MYO7A, поэтому при мутациях в гене MYO7A RPE65 частично локализован на свету и распадается быстрее, возможно, из-за его неправильной локализации, что дает правдоподобное объяснение его более низким уровням. После 50–60% фотозасвета сетчатка с мутацией в MYO7A демонстрирует повышенные уровни всех трансретиниловых эфиров на начальных стадиях темнового восстановления, что согласуется с дефицитом активности RPE65. Наконец, MYO7A и RPE65 совместно иммунопреципитируются из клеточного лизата RPE антителами против любого из белков, и два белка были локализованы в одних и тех же участках клетки, что указывает на их прямое или косвенное взаимодействие.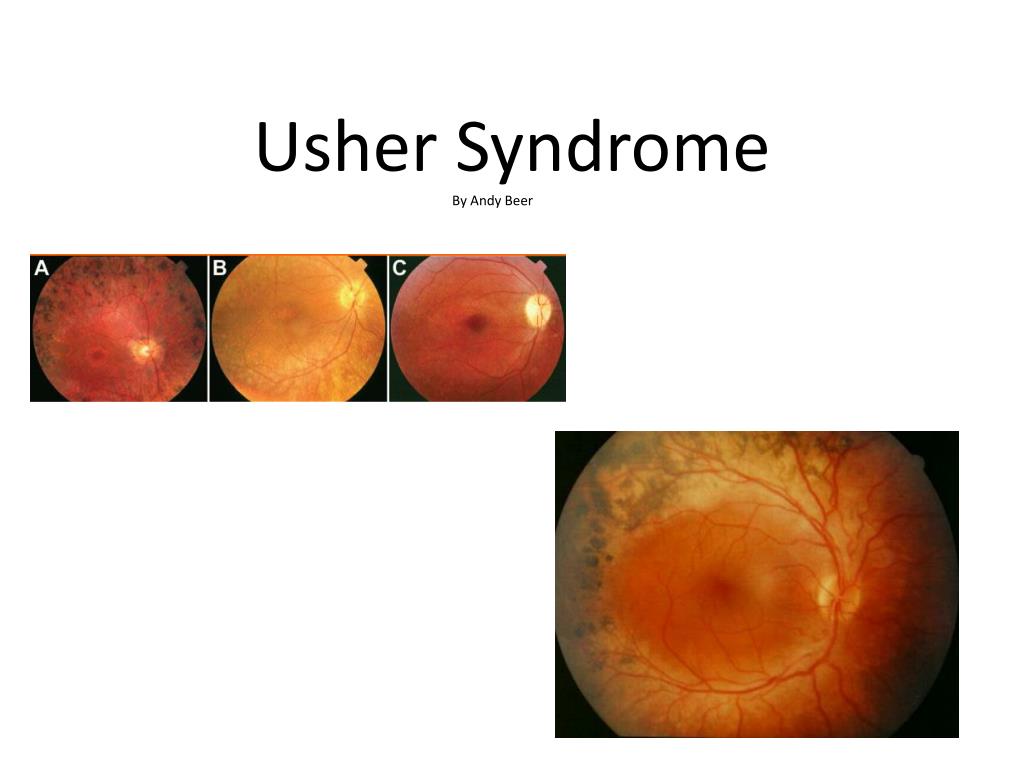 Вместе результаты подтверждают роль MYO7A в транслокации RPE65, иллюстрируя участие молекулярного мотора в пространственно-временной организации ретиноидного цикла в процессе фототрансдукции [21].
Вместе результаты подтверждают роль MYO7A в транслокации RPE65, иллюстрируя участие молекулярного мотора в пространственно-временной организации ретиноидного цикла в процессе фототрансдукции [21].
На основе этих знаний можно предположить, что зарегистрированный и разрешенный к применению на территории США препарат Luxturna (воретиген непарвовек), представляющий собой рабочую копию гена RPE65 на аденовирусном носителе, в будущем может показать свою эффективность при терапии MYO7A, ассоциированного с USh2. Также разрабатывается генно-терапевтический препарат UshStat (компания Sanofi совместно с Oxford biomedica) с доставкой правильно работающей копии гена MYO7A на лентивирусном носителе [22–25], однако продвижения в этой области происходят относительно медленно, в т. ч. в связи с повышенной иммуногенностью лентивирусной технологии доставки.
USh3A — ген, кодирующий белок ушерин, расположен на 1-й хромосоме и имеет 72 экзона. Ушерин содержит участки ламинина эндотелиального ростового фактора, домен пентаксина и множество участков фибронектина типа III, он находится в базальной мембране и важен для развития и поддержания гомеостаза внутреннего уха и сетчатки. Для этого гена было найдено несколько вариантов транскриптов, кодирующих разные изоформы белка. Мутации в гене USh3A являются наиболее частой причиной аутосомно-рецессивного несиндромального пигментного ретинита и USh3 в мировой статистике, однако в нашей группе пациентов в связи с особенностями формирования когорты (по критериям включения в группу принимались пациенты с наиболее серьезными нарушениями) большой процент составили пациенты с USh2. Никталопия и потеря периферического поля зрения в течение первых двух десятилетий, прогрессирующая с возрастом, наблюдались у пациентов с этим типом заболевания, хотя темпы прогрессирования варьировались с существенной внутри- и межсемейной фенотипической изменчивостью. Терапевтические подходы к коррекции недостаточной функции белка ушерина в связи с большим размером гена затруднены, однако разрабатываются различные подходы к доставке более длинных генов (так называемые наночастицы) и попытки замены частей гена (генная терапия мутаций в 13-м экзоне гена USh3A, разрабатываемая компанией ProQR), в т.
Ушерин содержит участки ламинина эндотелиального ростового фактора, домен пентаксина и множество участков фибронектина типа III, он находится в базальной мембране и важен для развития и поддержания гомеостаза внутреннего уха и сетчатки. Для этого гена было найдено несколько вариантов транскриптов, кодирующих разные изоформы белка. Мутации в гене USh3A являются наиболее частой причиной аутосомно-рецессивного несиндромального пигментного ретинита и USh3 в мировой статистике, однако в нашей группе пациентов в связи с особенностями формирования когорты (по критериям включения в группу принимались пациенты с наиболее серьезными нарушениями) большой процент составили пациенты с USh2. Никталопия и потеря периферического поля зрения в течение первых двух десятилетий, прогрессирующая с возрастом, наблюдались у пациентов с этим типом заболевания, хотя темпы прогрессирования варьировались с существенной внутри- и межсемейной фенотипической изменчивостью. Терапевтические подходы к коррекции недостаточной функции белка ушерина в связи с большим размером гена затруднены, однако разрабатываются различные подходы к доставке более длинных генов (так называемые наночастицы) и попытки замены частей гена (генная терапия мутаций в 13-м экзоне гена USh3A, разрабатываемая компанией ProQR), в т. ч. с применением технологии CRISPR [26]. Описание исследованной российской когорты пациентов с синдромом Ашера и частота мутаций в генах, наиболее частых при данном синдроме, представлены на рисунке 3 и в таблице 1.
ч. с применением технологии CRISPR [26]. Описание исследованной российской когорты пациентов с синдромом Ашера и частота мутаций в генах, наиболее частых при данном синдроме, представлены на рисунке 3 и в таблице 1.
PCDh25 — ген суперсемейства кадгеринов, кодирует один из подтипов протокадгерина, расположен на 10-й хромосоме и насчитывает 48 экзонов. Протокадгерины кодируют интегральные мембранные белки, которые обеспечивают кальций-зависимую клеточно-клеточную адгезию. Он играет существенную роль в поддержании нормальной функции сетчатки и кортиева органа. CDh33 вместе с PCDh25 образуют концевые звенья, которые соединяют стереоцилии, что крайне важно для поддержания жизнеспособности фоторецепторов и клеток кортиева органа. Мутации в этом гене приводят к потере слуха и развитию USh2. Характерен альтернативный сплайсинг, что приводит к появлению нескольких изоформ белка [27].
USh2C — ген ушерин 1С, расположен на 11-й хромосоме, имеет 29 экзонов. Этот ген кодирует белок-каркас, который участвует в сборке белковых комплексов. Белок содержит PDZ-домены, область сверхскрученной спирали с двойным сигналом ядерной локализации. Дефекты в этом гене являются причиной развития USh2C и несиндромальной нейросенсорной тугоухости аутосомно-рецессивного типа 18. Для этого гена было обнаружено множество вариантов транскриптов, кодирующих разные изоформы белка.
Этот ген кодирует белок-каркас, который участвует в сборке белковых комплексов. Белок содержит PDZ-домены, область сверхскрученной спирали с двойным сигналом ядерной локализации. Дефекты в этом гене являются причиной развития USh2C и несиндромальной нейросенсорной тугоухости аутосомно-рецессивного типа 18. Для этого гена было обнаружено множество вариантов транскриптов, кодирующих разные изоформы белка.
CDh33 — кадгерин-связанный белок 23-го подтипа, расположен на 10-й хромосоме, имеет 71 экзон. Этот ген также является членом суперсемейства кадгеринов. Белок с этого гена участвует в организации стереоцилий и формировании ресничек фоторецепторов. USh2D и несиндромальная аутосомно-рецессивная глухота DFNB12 вызваны аллельными мутациями этого кадгерин-подобного гена. Описаны альтернативные варианты сплайсинга, кодирующие разные изоформы этого белка [28].
Терапевтические подходы к коррекции функции генов USh2C, CDh33 и PCDh25 пока ограничиваются косвенными методиками — использованием аминогликозидов как ex vivo, так и in vivo для считывания преждевременных стоп-кодонов или нонсенс-мутаций [29]. Аминогликозиды в данном случае работают путем ослабления строгого механизма отбора рибосомой при синтезе белка из мРНК. Как при антибактериальном действии, когда они нарушают правильную генерацию полипептидов бактерий, так и в этом случае, расслабляя трансляцию, аминогликозид позволяет вставлять аминокислоту в область стоп-кодона вместо обрыва синтеза белка. Хотя эта аминокислота может быть не той аминокислотой, которая наблюдается в белке дикого типа, существует возможность восстановления правильной функции или по крайней мере лучшая функциональность получаемого белка ушерина, кадгерина или протокадгерина.
Аминогликозиды в данном случае работают путем ослабления строгого механизма отбора рибосомой при синтезе белка из мРНК. Как при антибактериальном действии, когда они нарушают правильную генерацию полипептидов бактерий, так и в этом случае, расслабляя трансляцию, аминогликозид позволяет вставлять аминокислоту в область стоп-кодона вместо обрыва синтеза белка. Хотя эта аминокислота может быть не той аминокислотой, которая наблюдается в белке дикого типа, существует возможность восстановления правильной функции или по крайней мере лучшая функциональность получаемого белка ушерина, кадгерина или протокадгерина.
Заключение
Авторами изложены результаты наблюдательного клинического исследования NCT03319524 и проведен подробный компьютерный анализ метаболома и интерактома, а также патогенетических путей развития USH в обследуемой когорте пациентов с обсуждением важности патогенетически направленных подходов к терапии и наиболее перспективных подходов к терапевтическому воздействию, в т. ч. с применением генной терапии.
ч. с применением генной терапии.
Финансирование/Funding
Исследование спонсировано БФ «Фонд поддержки слепоглухих «Со-единение» и АНО «Лаборатория Сенсор-Тех».
Autonomous non-commercial organization Laboratory “Sensor technology for deafblind” and Deaf-Blind Support Foundation “Con-nection”.
Сведения об авторах:
1Иванова Марианна Евгеньевна — к.м.н., руководитель, ORCID iD 0000-0002-1089-4293;
2Атарщиков Дмитрий Сергеевич — к.м.н., врач-офтальмолог, ORCID ID 0000-0003-4401-9099;
3Демчинский Андрей Михайлович — к.м.н., руководитель медицинских проектов, ORCID iD 0000-0002-1689-9394;
4Стрельников Владимир Викторович — д.б.н., заведующий лабораторией эпигенетики, ORCID iD 0000-0001-9283-902X;
5Бар Дебмала — PhD, начальник отдела исследований
и разработки, ORCID iD 0000-0002-2557-7768;
6Порядин Геннадий Васильевич — д.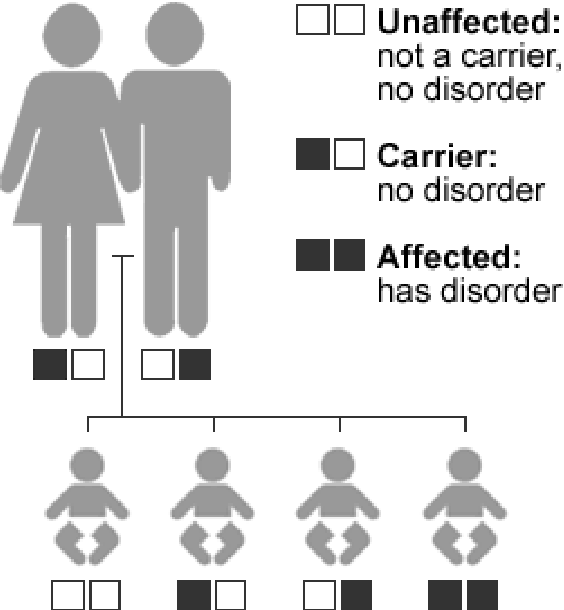 м.н., профессор, член-корр. РАН, почетный заведующий кафедрой патофизиологии, ORCID iD 0000-0003-2010-3296;
м.н., профессор, член-корр. РАН, почетный заведующий кафедрой патофизиологии, ORCID iD 0000-0003-2010-3296;
7Балашова Лариса Маратовна — д.м.н., профессор, руководитель, ORCID iD 0000-0001-9349-7092;
6Салмаси Жеан Мустафаевич — д.м.н., профессор, заведующий кафедрой патофизиологии, ORCID iD 0000-0001-8524-0019.
1НКЦ «Офтальмик», 125167, Россия, г. Москва, Ленинградский пр-т, д. 47/3–3.
2ФГБУ «ЦКБ с поликлиникой». 121359, Россия, г. Москва, ул. Маршала Тимошенко, д. 15.
3АНО «Лаборатория Сенсор-Тех». 115114, Россия, г. Москва, Павелецкая наб., д. 2, стр. 3.
4ФГБНУ “МГНЦ”. 115478, Россия, г. Москва, ул. Москворечье, д. 1.
5Центр геномики и прикладной генной технологии. 560032, Индия, г. Бангалор, ул. Чоланаяканахалли, д. 209.
6ФГБОУ ВО РНИМУ им.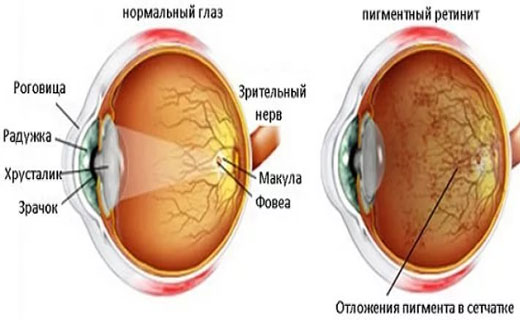 Н.И. Пирогова Минздрава России. 117513, Россия, г. Москва, ул. Островитянова, д. 1.
Н.И. Пирогова Минздрава России. 117513, Россия, г. Москва, ул. Островитянова, д. 1.
7НП «МНПЦПТ». 119034, Россия, г. Москва, ул. Пречистенка, д. 29/14.
Контактная информация: Иванова Марианна Евгеньевна, e-mail: [email protected]. Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах. Конфликт интересов: Иванова М.Е. является сотрудником НКЦ «Офтальмик». Остальные авторы заявляют об отсутствии конфликта интересов. Статья поступила 07.05.2019.
About the authors:
1Marianna E. Ivanova — MD, PhD, Head of CRO, ORCID iD 0000-0002-1089-4293;
2Dmitry S. Atarshchikov — MD, PhD, ophthalmologist, ORCID iD 0000-0003-4401-9099;
3Andrey M. Demchinsky— MD, PhD, Head of Medical Projects, ORCID iD 0000-0002-1689-9394;
4Vladimir V. Strelnikov — PhD, Head of Epigenetics Laboratory, ORCID iD 0000-0001-9283-902X;
Strelnikov — PhD, Head of Epigenetics Laboratory, ORCID iD 0000-0001-9283-902X;
5Debmalya Barh — PhD, Head of R&D Department, ORCID iD 0000-0002-2557-7768;
6Gennadiy V. Poryadin — MD, PhD, Professor, Corresponding Member of RAS, Honorary Head of Pathophysiology Department, ORCID iD 0000-0003-2010-3296;
7Larisa M. Balashova — MD, PhD, Professor, Head of the Center, ORCID iD 0000-0001-9349-7092;
6Jean M. Salmasi — MD, PhD, Professor, Head of Pathophysiology Department, ORCID iD 0000-0001-8524-0019.
1LLC “Oftalmic”. 47/3–3, Leningradsky Prospekt, Moscow,125167, Russian Federation.
2Central Clinical Hospital for Presidential Affairs. 15, Marshala Timoshenko str., Moscow,121359, Russian Federation.
3Autonomous nonprofit organization “Scientific and industrial laboratory “Sensor technology for deafblind”.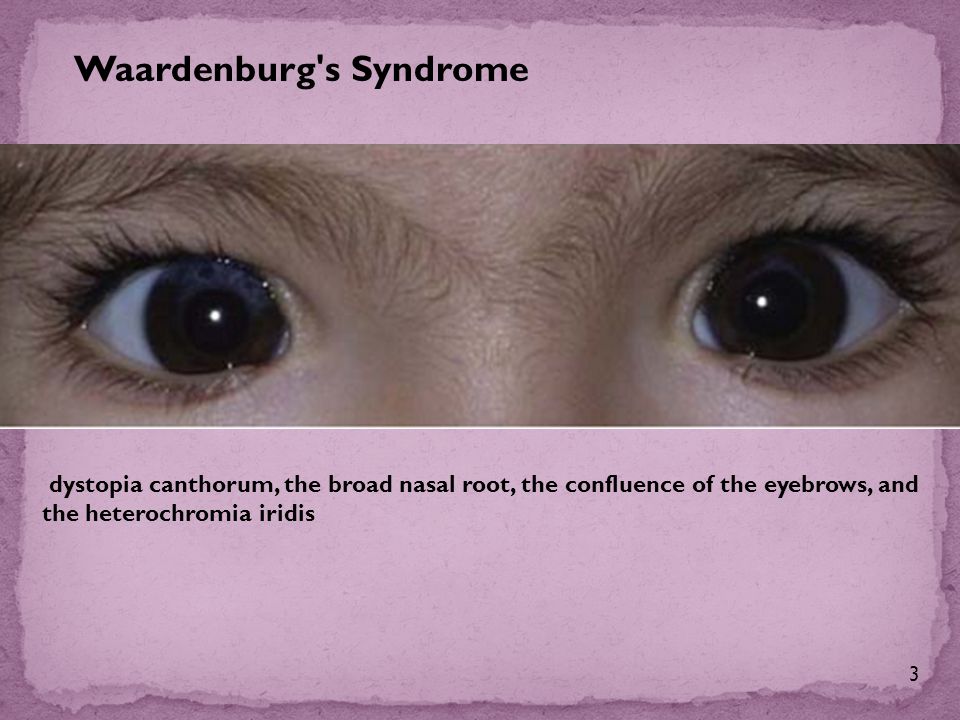 2, bld. 3, Paveletskaya naberezhnaya, Moscow, 115114, Russian Federation.
2, bld. 3, Paveletskaya naberezhnaya, Moscow, 115114, Russian Federation.
4Research Centre for Medical Genetics. 1, Moskvorechie str., Moscow, 115478, Russian Federation.
5Institute of Integrative Omics and Applied Biotechnology. 209, Cholanayakanahalli str., Bangalore, 560032, India.
6Pirogov Russian National Research Medical University. 1, Ostrovityanov str., Moscow, 117513, Russian Federation.
7Non-profit partnership International Scientific and Practical Center for the Proliferation of Tissues of Russia. 29/14, Prechistenka str., Moscow, 119034, Russian Federation.
Contact information: Marianna E. Ivanova, e-mail: [email protected]. Financial Disclosure: Marianna E. Ivanova is the collaborator of LLC “Oftalmic”. Other authors declare that there is no conflict of interests. Received 07.05.2019.
Синдром Ушера — NORD (Национальная организация по редким заболеваниям)
Синдром Ушера
NORD выражает благодарность Маргарет Кенна, доктору медицины, магистру здравоохранения, директору клинических исследований отделения отоларингологии и улучшения коммуникации Бостонской детской больницы и коалиции по лечению синдрома Ушера. , за помощь в подготовке этого отчета.
Подразделы синдрома Ушера
- Синдром Ушера типа 1
- Синдром Ушера типа 2
- Синдром Ушера типа 3
Признаки и симптомы
Причины
Синдром Ушера вызывается мутациями в определенных генах. До сих пор синдром Ушера был связан с мутациями по крайней мере в десяти генах:
Синдром Ушера типа 1: MYO7A (USh2B), USh2C, CDh33, PCDh25 (USh2F), SANS (USh2G) и, возможно, CIB2
Синдром Ушера 2 типа: USh3A, ADGRV1 (ранее назывался VLGR1 ) WHRN (DFNB31)
Синдром Ушера 3: USh4A (CLRN1) , HARS
Эти гены предоставляют инструкции для создания белков, участвующих в нормальном слухе, зрении и балансе. Некоторые из этих белков помогают специализированным клеткам, называемым волосковыми клетками, передавать звук от внутреннего уха к мозгу и воспринимать свет и цвет на сетчатке глаза. Функция некоторых белков, продуцируемых генами, ассоциированными с синдромом Ашера, неизвестна.
Некоторые из этих белков помогают специализированным клеткам, называемым волосковыми клетками, передавать звук от внутреннего уха к мозгу и воспринимать свет и цвет на сетчатке глаза. Функция некоторых белков, продуцируемых генами, ассоциированными с синдромом Ашера, неизвестна.
У некоторых людей с синдромом Ашера нет мутаций ни в одном из этих генов, поэтому, вероятно, существуют другие гены, связанные с этим заболеванием, которые еще не идентифицированы.
Все типы синдрома Ушера наследуются по аутосомно-рецессивному типу. Большинство генетических заболеваний определяются статусом двух копий гена, полученного от отца и матери. Рецессивные генетические нарушения возникают, когда человек наследует две копии аномального гена одного и того же признака, по одной от каждого родителя. Если человек наследует один нормальный ген и один ген болезни, он будет носителем болезни, но обычно не будет проявлять симптомов. Риск для двух родителей-носителей передать измененный ген и родить больного ребенка составляет 25% при каждой беременности. Риск рождения ребенка-носителя, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
Риск рождения ребенка-носителя, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
Родители, являющиеся близкими родственниками (кровнородственными), имеют более высокий шанс, чем неродственные родители, нести один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Пораженные группы населения
Синдром Ушера поражает от трех до десяти человек на 100 000 человек во всем мире. Больше, чем в среднем, число людей с синдромом Ушера было обнаружено среди евреев в Израиле, Берлине, Германии; Французские канадцы Луизианы; аргентинцы испанского происхождения; и нигерийские африканцы. USh4, самая редкая форма в большинстве популяций, составляет около 40% пациентов Usher в Финляндии. Синдром Ушера является наиболее распространенным генетическим заболеванием, сопровождающимся нарушением слуха и зрения. На синдром Usher 1 и 2 типов приходится примерно 10 процентов всех случаев глухоты от умеренной до полной у детей.
На синдром Usher 1 и 2 типов приходится примерно 10 процентов всех случаев глухоты от умеренной до полной у детей.
Диагностика
Синдром Ушера диагностируется при проверке слуха, равновесия и зрения. Слуховое (аудиологическое) обследование измеряет частоту и громкость звуков, которые может слышать человек. Электроретинограмма измеряет электрический отклик светочувствительных клеток сетчатки глаза. Обследование сетчатки проводится для наблюдения за сетчаткой и другими структурами в задней части глаза. Вестибулярную функцию (равновесие) можно оценить с помощью различных тестов, которые оценивают различные части системы равновесия. Генетическое тестирование клинически доступно для большинства генов, связанных с синдромом Ашера.
Стандартная терапия
Лечение
Лечение синдрома Ушера направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Такое лечение может потребовать скоординированных усилий группы медицинских работников, таких как педиатры или терапевты, специалисты по оценке и лечению нарушений слуха и равновесия (отоларингологи и аудиологи), врачи, специализирующиеся на диагностике и лечении заболеваний глаз (офтальмологи) и/или или других медицинских работников.
Нейросенсорная глухота должна оцениваться и изучаться варианты общения как можно раньше, чтобы дать ребенку прочную языковую базу. Слуховые аппараты или кохлеарные имплантаты принесут пользу большинству младенцев и детей с синдромом Ашера. Американский язык жестов можно использовать как средство общения. Люди, которые жестят визуально, часто переходят на тактильные знаки по мере ухудшения зрения. Раннее вмешательство важно для обеспечения того, чтобы дети с синдромом Ушера реализовали свой потенциал. Услуги, которые могут быть полезными, могут включать специальные услуги для детей с нейросенсорной глухотой или слепоглухотой и другие медицинские, социальные и/или профессиональные услуги.
В настоящее время не существует известного лекарства от RP, хотя исследователи работают над генетическими и другими методами лечения, чтобы восстановить или обратить вспять потерю зрения, связанную с RP, а также потерю слуха. Некоторые исследователи показали, что прием определенной суточной дозы витамина А может замедлить прогрессирование дегенерации сетчатки у некоторых людей с типичным РП и синдромом Ушера 2 типа. Некоторые эксперты рекомендуют взрослым пациентам с распространенными формами РП принимать 15 000 МЕ витамина А в день. palmitate под наблюдением офтальмологов, придерживаются регулярной сбалансированной диеты и избегают приема высоких доз витамина Е. Поскольку длительный прием высоких доз витамина А (например, превышающих 25 000 МЕ) может вызвать определенные побочные эффекты, такие как заболевания печени, пациенты должны регулярно находиться под наблюдением врача при приеме таких добавок. (Запасы витамина А в организме в основном хранятся в печени.) Очень важно, чтобы все пациенты с РП, рассматривающие такие добавки, проконсультировались со своими врачами для необходимой оценки, чтобы определить, является ли это целесообразным или нецелесообразным в их конкретном случае.
Некоторые эксперты рекомендуют взрослым пациентам с распространенными формами РП принимать 15 000 МЕ витамина А в день. palmitate под наблюдением офтальмологов, придерживаются регулярной сбалансированной диеты и избегают приема высоких доз витамина Е. Поскольку длительный прием высоких доз витамина А (например, превышающих 25 000 МЕ) может вызвать определенные побочные эффекты, такие как заболевания печени, пациенты должны регулярно находиться под наблюдением врача при приеме таких добавок. (Запасы витамина А в организме в основном хранятся в печени.) Очень важно, чтобы все пациенты с РП, рассматривающие такие добавки, проконсультировались со своими врачами для необходимой оценки, чтобы определить, является ли это целесообразным или нецелесообразным в их конкретном случае.
Людям с РП в сочетании с синдромом Ушера могут помочь средства для слабовидящих. Другое лечение синдрома Ушера является симптоматическим и поддерживающим. Агентства, которые предоставляют услуги людям с потерей слуха и зрения, могут быть полезными.
Генетическое консультирование рекомендуется больным людям и их семьям.
Исследовательская терапия
Информация о текущих клинических испытаниях размещена в Интернете на сайте www.clinicaltrials.gov. Все исследования, финансируемые правительством США, а некоторые из них поддерживаются частным сектором, публикуются на этом правительственном веб-сайте.
Для получения информации о клинических испытаниях, проводимых в Клиническом центре NIH в Бетесде, штат Мэриленд, обращайтесь в отдел набора пациентов NIH:
Бесплатный звонок: (800) 411-1222
Телетайп: (866) 411-1010
Электронная почта: [email защищено]
Некоторые текущие клинические испытания также размещены на следующей странице веб-сайта NORD:
https://rarediseases.org/for-patients-and-families/information-resources/news-patient-recruitment/
информация о клинических испытаниях, спонсируемых частными источниками, контакт:
www.centerwatch.com
Для получения информации о клинических испытаниях, проведенных в Европе, обращайтесь:
https://www. clinicaltrialsregister.eu/
clinicaltrialsregister.eu/
Ссылки
ИНТЕРНЕТ
Lentz J, Keats BJB. Usher Syndrome Type I. 10 декабря 1999 г. [обновлено 19 мая 2016 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2018 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1265/ По состоянию на 30 мая 2018 г.
Ленц Дж., Китс Б. Синдром Ашера Тип II. 10 декабря 1999 г. [Обновлено 21 июля 2016 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2018 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1341/ По состоянию на 30 мая 2018 г.
Синдром Ушера. Информационный центр NIDCD. Дата последнего обновления:
, 16 марта 2017 г. https://www.nidcd.nih.gov/health/usher-syndrome По состоянию на 30 мая 2018 г.
Синдром Ашера. Домашний справочник по генетике. Пересмотрено в ноябре 2017 г. https://ghr.nlm.nih.gov/condition/usher-syndrome. По состоянию на 30 мая 2018 г.
https://ghr.nlm.nih.gov/condition/usher-syndrome. По состоянию на 30 мая 2018 г.
МакКьюсик В.А., изд. Онлайн менделевское наследование у человека (OMIM). Балтимор. Доктор медицины: Университет Джона Хопкинса. https://www.omim.org/
Годы публикации
1989, 1990, 1993, 1996, 1997, 1998, 1999, 2000, 2001, 2005, 2018
Информация в базе данных редких заболеваний NORD предназначена для образовательных целей. только и не заменяет консультацию врача или другого квалифицированного медицинского работника.
Содержание веб-сайта и баз данных Национальной организации редких заболеваний (NORD) защищено авторским правом и не может быть воспроизведено, скопировано, загружено или распространено каким-либо образом в коммерческих или общественных целях без предварительного письменного разрешения и одобрения. от НОРД. Физические лица могут распечатать одну бумажную копию отдельного заболевания для личного использования при условии, что содержание не изменено и включает авторские права NORD.
Национальная организация редких заболеваний (NORD)
55 Kenosia Ave., Danbury CT 06810 • (203)744-0100
Что такое синдром Ашера? — Американская академия офтальмологии
Синдром Ушера – наиболее распространенное генетическое заболевание, поражающее как зрение, так и слух. Основными симптомами синдрома Ушера являются потеря слуха и потеря зрения из-за заболевания глаз, называемого пигментным ретинитом или РП.
Потеря зрения из-за RP может начаться в любом возрасте от раннего детства до подросткового возраста.
Кто подвержен риску синдрома Ушера?
Синдром Usher является рецессивным заболеванием. Это означает, что человек должен унаследовать изменение гена от обоих родителей. Человек, унаследовавший измененный ген от одного из родителей, не имеет синдрома, но является его носителем. Когда у двух носителей одного и того же гена синдрома Ушера есть общий ребенок, вероятность того, что у ребенка будет синдром Ушера, составляет один из четырех.
Основным визуальным симптомом синдрома Ашера является потеря зрения из-за пигментного ретинита (РП).
У людей с РП светочувствительные клетки сетчатки медленно дегенерируют (отмирают). Эти клетки называются палочками и колбочками. Это приводит к постепенной потере зрения. В большинстве форм RP первыми начинают умирать стержни. Это приводит к потере бокового и ночного зрения. Когда колбочки начинают отмирать, результатом является потеря восприятия цвета и центрального (чтения) зрения. Как правило, через много лет наступает слепота.
Синдром Usher влияет на зрение, слух и иногда на равновесие. Существует несколько типов синдрома Ушера в зависимости от задействованных генов.
Лучший способ точно диагностировать синдром Ашера — провести генетическое тестирование, которое может определить задействованные гены.
Оценка состояния глаз офтальмологом может включать:
- тест поля зрения для измерения периферического (бокового) зрения
- электроретинограмма (ЭРГ) для измерения реакции светочувствительных клеток глаза
- осмотр сетчатки для осмотра сетчатки и других структур в задней части глаза.

- оптическая когерентная томография (ОКТ) , которая позволяет получить изображение сетчатки в поперечном сечении.
Как лечится синдром Ушера?
В настоящее время не существует известного лекарства от синдрома Ашера или пигментного ретинита (РП). Знание генов, ответственных за синдром Ашера, может помочь вам найти продолжающееся испытание генной терапии и, в конечном итоге, одобренную терапию. Лучшее лечение включает раннюю диагностику, чтобы образовательные программы могли начаться как можно раньше, в зависимости от серьезности потери зрения, возраста и способностей ребенка.
Лечение может включать:
- инструкции по чтению шрифтом Брайля
- и обучение использованию устройств и методов для слабовидящих.
Некоторые исследования показали, что витамин А может замедлять прогрессирование некоторых форм РП, но есть опасения, что высокое потребление витамина А может привести к ухудшению других глазных заболеваний.