
Митохондриальный синдром: Митохондриальные заболевания, комплексная диагностика: митохондриальная ДНК, ч. м. (Mitochondrial Diseases, multiplex mutations detection assay)

заболевания митохондриального типа
заболевания митохондриального типа
14.11.2022
Количество просмотров 2639
Митохондриальные заболевания — это заболевания, при которых имеется дефект в митохондриях, обычно имеющий генетическую причину. Функциональные нарушения митохондрий в первую очередь затрагивают мышечные клетки, так как они имеют высокую потребность в энергии. Может возникнуть митохондриальная миопатия (заболевание мышц). Но могут быть затронуты и другие клетки и ткани, такие как нервная система, глаза и внутреннее ухо. Встречаются также расстройства желудочно-кишечного тракта, печени или поджелудочной железы и т.д.
Английское название:
Mitochondrial diseases
Синонимы:
Наследственные заболевания
См. также:
Аутоиммунные заболевания
Генетические заболевания
Содержание
- Клинические симптомы
- Заболевания
- Лечение
Поэтому заболевания митохондрий часто представляют собой так называемые полисистемные заболевания, при которых поражаются различные органы. Митохондриальное заболевание может возникать как в детстве, так и во взрослом возрасте, клинические проявления могут меняться с течением времени.
Митохондриальное заболевание может возникать как в детстве, так и во взрослом возрасте, клинические проявления могут меняться с течением времени.
Клинические симптомы
Дефекты в митохондриях приводят к нарушению клеточного энергетического обмена и сегодня играют важную роль в педиатрической и неврологической клинике, хотя распространенность этих заболеваний еще трудно оценить (примерно 1:5000).
Дефекты дыхательной цепи относятся к классическим и наиболее изученным митохондриальным заболеваниям. Первичные дефекты дыхательной цепи затрагивают либо структурные подразделения самой дыхательной цепи, либо факторы более высокого уровня. Они вызывают первичный дефицит клеточной энергии (дефицит АТФ ) и во многих случаях приводят к прогрессирующим нейродегенеративным заболеваниям. Дефекты дыхательной цепи клинически крайне гетерогенны. Наиболее часто поражаются органы с высоким энергообменом, т.е. ЦНС , скелетные мышцы и сердце. Поэтому частыми симптомами со стороны мускулатуры являются мышечные боли при нагрузке, а также мышечная слабость (парез) и мышечная атрофия. Частыми дополнительными симптомами являются сахарный диабет, слепота (ретинопатия), невропатия, глухота, нарушения сердечного ритма и другие.
Частыми дополнительными симптомами являются сахарный диабет, слепота (ретинопатия), невропатия, глухота, нарушения сердечного ритма и другие.
Заболевания
В последнее десятилетие описано более 200 различных мутаций митохондриальных ДНК и дефектов многочисленных ядерных генов, которые вызывают первичные дефекты дыхательной цепи и иногда встречаются спорадически, но также могут наследоваться по материнскому, аутосомно-рецессивному, аутосомно-доминантному наследованию. Таким образом, клиническая и генетическая гетерогенность представляет особую проблему как для диагностики, так и для генетического консультирования.
Здесь кратко представлены некоторые митохондриальные заболевания, которые в основном проявляются симптомами в мышцах:
При MERRF пациенты страдают от таких симптомов, как миоклоническая и тонико-клоническая эпилепсия, митохондриальная миопатия, нейросенсорная глухота, атаксия и деменция. Типичным гистологическим коррелятом этого митохондриального заболевания в мышцах является «рваное красное волокно» (RRF). Наиболее частой причиной синдрома MERRF является мутация 8344A>G в гене мтДНК- тРНК -Lys. Существуют также прогрессирующие миоклонус-эпилепсии с RRF или без него, которые могут быть связаны с другими мутациями в мтДНК (например, ND5, другими генами тРНК). Аутосомно-наследственные мутации гена POLG1, кодируемого ядром, также были описаны у пациентов с типичным синдромом MERRF. У этих пациентов часто встречаются множественные делеции мтДНК в мышечной ДНК.
Наиболее частой причиной синдрома MERRF является мутация 8344A>G в гене мтДНК- тРНК -Lys. Существуют также прогрессирующие миоклонус-эпилепсии с RRF или без него, которые могут быть связаны с другими мутациями в мтДНК (например, ND5, другими генами тРНК). Аутосомно-наследственные мутации гена POLG1, кодируемого ядром, также были описаны у пациентов с типичным синдромом MERRF. У этих пациентов часто встречаются множественные делеции мтДНК в мышечной ДНК.
Изолированные глазные симптомы (паралич глазных мышц и опущение век) обнаруживаются при CPEO. Если с ним связаны другие симптомы, говорят о CPEO.+ вплоть до наиболее тяжелой формы синдрома Кернса-Сейра, который помимо глазной миопатии с ретинопатией, молочнокислым ацидозом, нейросенсорной глухотой, атаксией, кардиомиопатией могут быть связаны нарушения проводимости, повышение концентрации белка в ликворе и деменция. Начало заболевания вариабельно (от детского до старческого возраста). Эти клинические картины часто вызваны единичными делециями в митохондриальной ДНК. Делеции могут затрагивать участки митохондриальной ДНК разного размера; они возникают спонтанно за немногими исключениями.
Делеции могут затрагивать участки митохондриальной ДНК разного размера; они возникают спонтанно за немногими исключениями.
-
Рабдомиолиз.
Рабдомиолиз — распространенный клинический синдром, обусловленный острым некрозом мышечных волокон и выбросом мышечных ферментов в кровь, что приводит к миоглобинурии. Миоглобинурия может вызвать острую почечную недостаточность у 15% больных. Генетические заболевания, токсины, мышечная перегрузка или воспалительные процессы могут вызвать рабдомиолиз. Генетический фон рабдомиолиза очень неоднороден. У пациентов были описаны аутосомно-рецессивные мутации в гене карнитин-пальмитоилтрансферазы (CPT2) или в гене мышечной гликогенфосфорилазы (PYGM). Другой генетической причиной, обнаруживаемой у пациентов с митохондриальной миопатией и рабдомиолизом, являются мутации мтДНК в генах cytb, ND и тРНК.
-
Синдром Ли.
Синдром Ли или подострая некротизирующая энцефаломиелопатия названа по имени патологоанатома Д. Ли, впервые описавшего типичную морфологию головного мозга с симметричными базальными ганглиями и некрозом ствола в 1951 г. Синдром Ли является одним из наиболее распространенных митохондриальных заболеваний детского возраста. Примерно в 70% случаев СЛ связан с дефектами первичной дыхательной цепи, а еще в 15-20% — с недостаточностью пируватдегидрогеназы. В дополнение к типичному синдрому Ли существует ряд заболеваний дыхательной цепи, которые связаны с поражением базальных ганглиев, а также белого вещества. Имеются, например, явления демиелинизации, а также мозговые и мозжечковые атрофии. Также может быть демиелинизация периферических нервов с последующей нейрогенной атрофией мышц. Помимо изменений ЦНС, часто поражаются и другие органы, такие как сердце, печень, почки и др. Поскольку может быть выявлено большое количество причинных генных мутаций, возможна целенаправленная молекулярно-генетическая пренатальная диагностика.
Ли, впервые описавшего типичную морфологию головного мозга с симметричными базальными ганглиями и некрозом ствола в 1951 г. Синдром Ли является одним из наиболее распространенных митохондриальных заболеваний детского возраста. Примерно в 70% случаев СЛ связан с дефектами первичной дыхательной цепи, а еще в 15-20% — с недостаточностью пируватдегидрогеназы. В дополнение к типичному синдрому Ли существует ряд заболеваний дыхательной цепи, которые связаны с поражением базальных ганглиев, а также белого вещества. Имеются, например, явления демиелинизации, а также мозговые и мозжечковые атрофии. Также может быть демиелинизация периферических нервов с последующей нейрогенной атрофией мышц. Помимо изменений ЦНС, часто поражаются и другие органы, такие как сердце, печень, почки и др. Поскольку может быть выявлено большое количество причинных генных мутаций, возможна целенаправленная молекулярно-генетическая пренатальная диагностика.
Лечение
Причина митохондриальных заболеваний, т. е. дефекты митохондриальных или ядерных генов, пока не поддается лечению. Однако для многих заболеваний существуют варианты симптоматического лечения.
е. дефекты митохондриальных или ядерных генов, пока не поддается лечению. Однако для многих заболеваний существуют варианты симптоматического лечения.
Например, аэробные упражнения могут улучшить мышечную силу у пациентов со спорадическими мутациями мтДНК (выявляемыми только в мышечной ДНК). Различные симптомы и осложнения (эпилепсия, сахарный диабет, инсульты, сердечные аритмии, гормональные нарушения и др.) можно лечить или предотвращать с помощью медикаментов или других мер.
Интересуетесь антивозрастной
и превентивной медициной?
Узнайте больше на
обучающих программах Anti-Age Expert
Чтобы стать лучшим — учитесь
у лучших!
Эксперты со всего мира станут вашими наставниками
на пути изучение Anti-Age Expert. Подробнее
Список литературы
- Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing.
 N Engl J Med. 2014;
N Engl J Med. 2014; - Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;
- Chinnery PF, DiMauro S, Shanske S, Schon EA, Zeviani M, Mariotti C, Carrara F, Lombes A, Laforet P, Ogier H, Jaksch M, Lochmüller H, Horvath R, Deschauer M, Thorburn DR, Bindoff LA, Poulton J, Taylor RW, Matthews JN, Turnbull DM. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;
- Hellebrekers DM, Wolfe R, Hendrickx AT, de Coo IF, de Die CE, Geraedts JP, Chinnery PF, Smeets HJ. PGD and heteroplasmic mitochondrial DNA point mutations: a systematic review estimating the chance of healthy offspring. Hum Reprod Update. 2012.
Статьи по теме
14.04.2021
6 перспективных технологий в борьбе со старением
На протяжении десятилетий ученые работали над тем, чтобы подобрать ключи к сохранению молодости и здоровья. И в 21 веке борьба со старением только набирает обороты.
И в 21 веке борьба со старением только набирает обороты.
30.03.2021
Митохондриальная теория старения
Старение — сложный и многоэтапный процесс, объяснить который пытаются многие научные гипотезы. Например, митохондриальная теория. Она связывает старение с изменениями в функции митохондрий, энергетических центров клетки.
02.04.2021
Эпигенетические часы: потенциал изучения
Паспортный возраст – единица условная, которая едва ли расскажет о состоянии здоровья человека. Ведь даже ровесники обычно выглядят и чувствуют себя по-разному, а значит, и процессы старения у всех протекают с разной скоростью.
Развернутая диагностика митохондриальных заболеваний (MELAS, MERRF, прогрессирующая офтальмоплегия, с-м Кернса-Сейра, нейропатия Лебера)
Артикул: 10258
Анализ доступен:
Медицинский центр на площади Стачек, 5Медицинский центр на Богатырском пр. , 4Лабораторный терминал на пр. Александровской Фермы, 8Лабораторный терминал на пр. Наставников, 36к2Лабораторный терминал на ул. Будапештская, 6Медицинский центр на Пулковском шоссе, 28А Медицинский центр на Кондратьевском пр., 62к3Медицинский центр на пр. Просвещения, 14к4Медицинский центр на ул. Моисеенко, 5Лабораторный терминал на ул. Олеко Дундича, 8, к. 2Лабораторный терминал на ул. Пестеля, 25АМедицинский центр на Ленинском пр., 88Лабораторный терминал на ул. Турку, 5/13г. Санкт-Петербург, Выездная службаМедицинский центр на ул. Савушкина, д. 14
, 4Лабораторный терминал на пр. Александровской Фермы, 8Лабораторный терминал на пр. Наставников, 36к2Лабораторный терминал на ул. Будапештская, 6Медицинский центр на Пулковском шоссе, 28А Медицинский центр на Кондратьевском пр., 62к3Медицинский центр на пр. Просвещения, 14к4Медицинский центр на ул. Моисеенко, 5Лабораторный терминал на ул. Олеко Дундича, 8, к. 2Лабораторный терминал на ул. Пестеля, 25АМедицинский центр на Ленинском пр., 88Лабораторный терминал на ул. Турку, 5/13г. Санкт-Петербург, Выездная службаМедицинский центр на ул. Савушкина, д. 14
Цена:
6 635 ₽
В корзину
Вы можете заказать бесплатную услугу по сдаче анализа на дому
Делеции и дупликации митохондриальной ДНК (мтДНК), а также точечные мутации 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C могут вызывать следующие заболевания и состояния: MELAS синдром (митохондриальная миопатия, энцефалопатия, лактат-ациодоз, инсультоподобные эпизоды), MERRF синдром (миоклоническая эпилепсия, ассоциированная порванными красными волокнами), прогрессирующая наружная офтальмоплегия, синдром Кернса — Сейра, наследственная оптическая нейропатия Лебера. Нужно отметить, что клинические проявления данных заболеваний могут сильно варьировать и быть очень неспецифичными, а исследуемые мутации могут вызывать симптоматику, которая не вписывается в приведенные синдромы.
Нужно отметить, что клинические проявления данных заболеваний могут сильно варьировать и быть очень неспецифичными, а исследуемые мутации могут вызывать симптоматику, которая не вписывается в приведенные синдромы.
Синонимы: Митохондриальные заболевания, комплексная диагностика/ Mitochondrial Diseases, multiplex mutations detection assay
Оборудование: A/E/H
* актуальную информацию уточняйте в контактном центре по тел. 8 (812) 600-42-00
Специальной подготовки к исследованию не требуется.
Похожие анализы
Молекулярно-генетический тест диагностики дефицита мевалонат-киназы (MVK) и TRAPS-синдрома
до 9 дней
от 8 400 ₽
В корзину
Стандартный анализ наследственной мутации RET (тестирование для родственника пациента с уже выявленной RET мутацией)
до 30 дней
от 3 075 ₽
В корзину
Генетическая диагностика альфа-талассемии (мутации в гене HBA)
до 9 дней
от 9 135 ₽
В корзину
Генетическая диагностика бета-талассемии и гемоглобинопатий (мутации в гене HBB)
до 9 дней
от 9 135 ₽
В корзину
Таргетное секвенирование ДНК- анализ последовательности гена MEN1
45 дней (возможно увеличение срока до 60 дней)
от 20 895 ₽
В корзину
Таргетное секвенирование ДНК — анализ последовательности гена RET
45 дней (возможно увеличение срока до 60 дней)
от 20 895 ₽
В корзину
HLA B51 типирование (болезнь Бехчета)
до 9 дней
от 2 165 ₽
В корзину
Развернутая диагностика митохондриальных заболеваний (MELAS, MERRF, прогрессирующая офтальмоплегия, с-м Кернса-Сейра, нейропатия Лебера)
до 9 дней
от 6 635 ₽
В корзину
Молекулярное генотипирование числа Х-хромосом (с-мы Клайнфельтера, Тернера, 3ой Х)
до 9 дней
от 3 950 ₽
В корзину
Определение предэкспансии при первичной яичниковой недостаточности (в гене FMR1)
до 9 дней
от 5 460 ₽
В корзину
Генодиагностика болезни Шарко-Мари-Тута 1А и наследственной нейропатии с подверженностью к параличу от сдавления (PMP22)
до 9 дней
от 5 250 ₽
В корзину
Генодиагностика мышечной дистрофии Дюшенна и Беккера (DMD)
до 9 дней
от 6 070 ₽
В корзину
Генодиагностика спинальной мышечной атрофии (SMN1,SMN2)
до 9 дней
от 7 350 ₽
В корзину
Анализ доступен в этих центрах:
Медицинский центр на Богатырском пр. , 4
, 4
Медицинский центр на площади Стачек, 5
Медицинский центр на ул. Моисеенко, 5
Медицинский центр на пр. Просвещения, 14к4
Медицинский центр на ул. Савушкина, д. 14
14
Лабораторный терминал на ул. Турку, 5/13
Лабораторный терминал на ул. Пестеля, 25А
Лабораторный терминал на ул. Олеко Дундича, 8, к. 2
Лабораторный терминал на ул. Будапештская, 6
Будапештская, 6
Медицинский центр на Пулковском шоссе, 28А
Лабораторный терминал на пр. Наставников, 36к2
Лабораторный терминал на пр. Александровской Фермы, 8
Медицинский центр на Ленинском пр.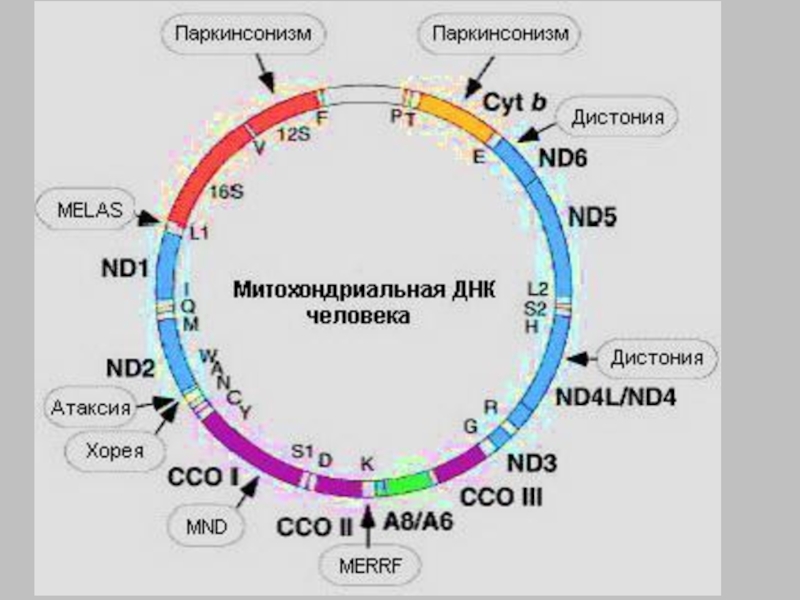 , 88
, 88
Медицинский центр на Кондратьевском пр., 62к3
Клиника ОРТОКРОСС на 5-ой линии В. О., д. 8А (официальный партнёр)
Лабораторный терминал на Кронверкском пр., 31 (официальный партнёр)
Клиника “ПулковоСтом” на Пулковском шоссе, д. 26, к.6. (официальный партнёр)
26, к.6. (официальный партнёр)
Лабораторный терминал на ул. Савушкина, 124 (официальный партнёр)
Митохондриальная болезнь | UMDF
Что такое митохондриальная болезнь?
Когда митохондрии не могут преобразовывать пищу и кислород в энергию для поддержания жизни, происходит повреждение клеток и даже их гибель. Когда этот процесс повторяется во всем организме, системы органов начинают давать сбои и даже перестают функционировать.
Почему Митохондрии Неисправны?
Нам еще многое предстоит открыть, но это то, что мы знаем. Митохондриальная болезнь является наследственным заболеванием. На ваши митохондрии также могут влиять другие генетические нарушения и факторы окружающей среды. Вы можете узнать больше о биологии митохондриальных заболеваний здесь .
Вы можете узнать больше о биологии митохондриальных заболеваний здесь .
Каждые 30 минут рождается ребенок, у которого к 10 годам разовьется митохондриальное заболевание.
Как митохондриальное заболевание влияет на организм
наиболее подвержены митохондриальным заболеваниям. Пораженный человек может проявлять спектр симптомов.
задержка развития, деменция, мигрень, аутистические черты, судороги, инсульт, атипичный церебральный паралич, нарушение обучаемости
слабость/отказ, судороги, рефлюкс, рвота, запор, диарея, гипотония, нарушение моторики креативная недостаточность, недостаточность паращитовидных желез
недостаточность почечной трубки
дефекты, блокада, кардиомиопатия
низкий уровень сахара в крови, печеночная недостаточность
потеря зрения, птоз, атрофия зрительного нерва, косоглазие, офтальмоплегия пигментный тинит
потеря слуха
неспособность набрать вес, усталость, низкий рост, необъяснимая рвота, проблемы с дыханием
Существует много типов митохондриальных заболеваний. Каждое расстройство вызывает спектр аномалий, которые могут сбивать с толку как пациентов, так и врачей.
Каждое расстройство вызывает спектр аномалий, которые могут сбивать с толку как пациентов, так и врачей.
Узнать больше
3+ нарушение работы систем органов является тревожным сигналом для митохондриального заболевания.
Узнайте, имеете ли вы право на бесплатный генетический тест.
Начало работы →
Прочтите статью «Диагностика и лечение митохондриальных заболеваний».
Просмотреть документ →
UMDF ведет список более чем 200 врачей, лечащих и исследующих митохондриальные заболевания.
Поиск поставщика услуг →
Сколько людей страдают митохондриальными заболеваниями?
Точное число людей, страдающих митохондриальным заболеванием, определить трудно, потому что многим из них часто ставится неправильный диагноз. Один из 5000 человек имеет генетическое митохондриальное заболевание.
Могут ли взрослые болеть митохондриальными заболеваниями?
Хотя это заболевание в основном поражает детей, начало заболевания у взрослых становится все более распространенным. Сам процесс старения может быть результатом ухудшения функции митохондрий. Существует широкий спектр метаболических, наследственных и приобретенных нарушений у взрослых, которые могут быть связаны с аномальной функцией митохондрий.
Сам процесс старения может быть результатом ухудшения функции митохондрий. Существует широкий спектр метаболических, наследственных и приобретенных нарушений у взрослых, которые могут быть связаны с аномальной функцией митохондрий.
Когда человек с митохондриальным заболеванием относится к группе высокого риска?
Как дети, так и взрослые подвержены высокому риску неврологического и органного повреждения во время повышенных нагрузок на организм. Это включает в себя во время и в течение двух недель после болезни, голодание, обезвоживание, хирургическое вмешательство, анестезию и внутривенное введение антибиотиков.
Каков прогноз для лиц, страдающих митохондриальными заболеваниями?
На этот вопрос сложно ответить, поскольку прогноз зависит от множества критериев. Некоторые пострадавшие дети и взрослые живут вполне нормальной жизнью. В других случаях больные дети могут не дожить до подросткового возраста. Начало во взрослом возрасте может привести к резкому изменению образа жизни и физическим изменениям за короткий промежуток времени.
Как UMDF поддерживает исследования по поиску лекарства?
UMDF финансирует лучшую науку по всему миру, в том числе миллионы долларов на исследования для улучшения методов лечения и лечения. Наша программа исследовательских грантов и 9Программа ускорителей 0022 поддерживает лучшие умы митохондриальной медицины.
Получите образование в Университете Мито
Получите доступ к видео, статьям и ресурсам, чтобы лучше ориентироваться в своем путешествии.
Посетите Mito U
В то время как мы боремся за поиск лекарства от митохондриального заболевания, существуют некоторые методы лечения, витамины и пищевые добавки, которые помогут облегчить симптомы и замедлить прогрессирование заболевания. Однако эффективность современных методов лечения сильно различается. Наша лучшая надежда на улучшение ухода за пациентами — это финансирование исследований митохондриальных заболеваний и клинических испытаний.
От 1000 до 4000 детей в США ежегодно рождаются с митохондриальными заболеваниями.
Присоединяйтесь к mitoSHARE: Реестр митохондриальных заболеваний
Помогите продвинуться вперед в направлении улучшения диагностики, лечения и лечения.
Зарегистрируйтесь
Зарегистрируйтесь в клиническом испытании, чтобы помочь в поиске лекарства
Узнайте, имеете ли вы право участвовать в клинических исследованиях.
Начало работы
Привлечение средств на исследования для поиска более эффективных методов лечения
Финансовые пожертвования программам поддержки UMDF для улучшения лечения митохондриальных заболеваний.
Узнать больше
Обзор первичных митохондриальных заболеваний — GeneReviews®
Barragán-Campos HM, Vallee JN, Lo D, Barrera-Ramirez CF, Argote-Greene M, Sanchez-Guerrero J, Estanol B, Guillevin R, Chiras J Результаты магнитно-резонансной томографии головного мозга у пациентов с митохондриальными цитопатиями. Арх Нейрол. 2005; 62: 737–42. [PubMed: 15883260]
Bisecker LG, Green RC. Диагностическое клиническое секвенирование генома и экзома. N Engl J Med. 2014; 370:2418–25. [В паблике: 24941179]
Диагностическое клиническое секвенирование генома и экзома. N Engl J Med. 2014; 370:2418–25. [В паблике: 24941179]
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Спастическая параплегия и Нарушение OXPHOS, вызванное мутациями параплегина, митохондриальной металлопротеазы, кодируемой ядром. Клетка. 1998; 93: 973–83. [PubMed: 9635427]
Чиннери П.Ф., ДиМауро С., Шанске С., Шон Э.А., Зевиани М., Мариотти С., Каррара Ф., Ломбес А., Лафоре П., Ожье Х., Якш М., Лохмюллер Х., Хорват Р., Дешауэр М. , Thorburn DR, Bindoff LA, Poulton J, Taylor RW, Matthews JN, Turnbull DM. Риск развития нарушения делеции митохондриальной ДНК. Ланцет. 2004;364:592–6. [PubMed: 15313359]
Hellebrekers DM, Wolfe R, Hendrickx AT, de Coo IF, de Die CE, Geraedts JP, Chinnery PF, Smeets HJ. PGD и точечные мутации гетероплазматической митохондриальной ДНК: систематический обзор, оценивающий вероятность здорового потомства. Обновление воспроизведения гула. 2012;18:341–9. [PubMed: 22456975]
Обновление воспроизведения гула. 2012;18:341–9. [PubMed: 22456975]
Холт И.Дж., Хардинг А.Е., Морган-Хьюз Дж.А. Делеции митохондриальной ДНК мышц у пациентов с митохондриальными миопатиями. Природа. 1988; 331: 717–9. [В паблике: 2830540]
Холт И.Дж., Хардинг А.Е., Петти Р.К., Морган-Хьюз Дж.А. Новое митохондриальное заболевание, связанное с гетероплазмией митохондриальной ДНК. Am J Hum Genet. 1990; 46: 428–33. [Бесплатная статья PMC: PMC1683641] [PubMed: 2137962]
Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Арнадоттир Г.А., Хельгасон Э.А., Хельгасон Х., Гилфасон А., Йонасдоттир А., Йонасдоттир А., Рафнар Т., Фригг М., Стейси С.Н., Т. Магнуссон О., Торстейнсдоттир У., Массон Г., Конг А., Халлдорссон Б.В., Хельгасон А., Гудбьяртссон Д.Ф., Стефанссон К. Влияние родителей на мутации de novo в зародышевой линии человека у 1548 троек из Исландии. Природа. 2017;549: 519–22. [PubMed: 28959963]
Природа. 2017;549: 519–22. [PubMed: 28959963]
Кауфманн П., Энгельстад К., Вей Ю., Куликова Р., Оскуи М., Баттиста В., Кенигсбергер Д.Ю., Паскуаль Дж.М., Сано М., Хирано М., ДиМауро С., Шунгу Д.К., Мао Х, Де Виво ОКРУГ КОЛУМБИЯ. Протеиновые фенотипические особенности мутации митохондриальной ДНК A3243G. Арх Нейрол. 2009;66:85–91. [PubMed: 19139304]
Леонард СП, Шапира АВХ. Нарушения митохондриальной дыхательной цепи I: дефекты митохондриальной ДНК. Ланцет. 2000а; 355: 299–304. [PubMed: 10675086]
Леонард СП, Шапира АВХ. Митохондриальные расстройства дыхательной цепи II: нейродегенеративные расстройства и дефекты ядерных генов. Ланцет. 2000б; 355:389–94. [PubMed: 10665569]
Lieber DS, Calvo SE, Shanahan K, Slate NG, Liu S, Hershman SG, Gold NB, Chapman BA, Thorburn DR, Berry GT, Schmahmann JD, Borowsky ML, Mueller DM, Sims KB , Мута ВК. Целевое секвенирование экзома при подозрении на митохондриальные нарушения. Неврология. 2013; 80: 1762–70. [Бесплатная статья PMC: PMC3719425] [PubMed: 23596069]
2013; 80: 1762–70. [Бесплатная статья PMC: PMC3719425] [PubMed: 23596069]
Луценко С., Купер М.Ю. Локализация белкового продукта болезни Вильсона в митохондриях. Proc Natl Acad Sci U S A. 1998; 95:6004–9. [Бесплатная статья PMC: PMC27575] [PubMed: 9600907]
Macmillan C, Lach B, Shoubridge EA. Вариабельное распределение мутантных митохондриальных ДНК (тРНК (Leu[3243])) в тканях симптоматических родственников с MELAS: роль митотической сегрегации. Неврология. 1993; 43: 1586–90. [PubMed: 8351017]
Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, Rahman S, Hanna MG, McFarland R. Когортное исследование пациентов с митохондриальными заболеваниями MRC Великобритании: клинические фенотипы, связанные с мутацией m.3243A>G- — последствия для диагностики и лечения. J Neurol Нейрохирург Психиатрия. 2013; 84: 936–8. [PubMed: 23355809]
Парих С., Гольдштейн А., Караа А., Кениг М.К., Ансельм И., Брюнель-Гиттон С. , Христодулу Дж., Коэн Б.Х., Диммок Д., Эннс Г.М., Фальк М.Дж., Фейгенбаум А., Фрай Р.Е., Ганеш Дж., Гриземер Д., Хаас Р., Хорват Р., Корсон М., Крюер М.С., Манкузо М., МакКормак С., Рабуассон М.Дж., Раймшизель Т., Сальваринова Р., Сането Р.П., Скалья Ф., Шоффнер Дж., Стакпул П.В., Сью К.М., Тарнопольски М. , Ван Карнебек С., Вулф Л.А., Каннингем З.З., Рахман С., Чиннери П.Ф. Стандарты ухода за пациентами при первичном митохондриальном заболевании: консенсусное заявление Общества митохондриальной медицины. Генет Мед. 2017;19:1380. [Бесплатная статья PMC: PMC7804217] [PubMed: 28749475]
, Христодулу Дж., Коэн Б.Х., Диммок Д., Эннс Г.М., Фальк М.Дж., Фейгенбаум А., Фрай Р.Е., Ганеш Дж., Гриземер Д., Хаас Р., Хорват Р., Корсон М., Крюер М.С., Манкузо М., МакКормак С., Рабуассон М.Дж., Раймшизель Т., Сальваринова Р., Сането Р.П., Скалья Ф., Шоффнер Дж., Стакпул П.В., Сью К.М., Тарнопольски М. , Ван Карнебек С., Вулф Л.А., Каннингем З.З., Рахман С., Чиннери П.Ф. Стандарты ухода за пациентами при первичном митохондриальном заболевании: консенсусное заявление Общества митохондриальной медицины. Генет Мед. 2017;19:1380. [Бесплатная статья PMC: PMC7804217] [PubMed: 28749475]
Poulton J, Turnbull DM. 74-й международный семинар ENMC: митохондриальные болезни 19-20 ноября 1999 г., Наарден, Нидерланды. Нервно-мышечное расстройство. 2000; 10:460–2. [PubMed: 10899455]
Ретиг А., де Лонле П., Кретьен Д., Фури Ф., Кениг М., Сиди Д., Мюнних А., Растин П. Дефицит аконитазы и митохондриального железо-серного белка при атаксии Фридрейха. Нат Жене. 1997; 17: 215–7.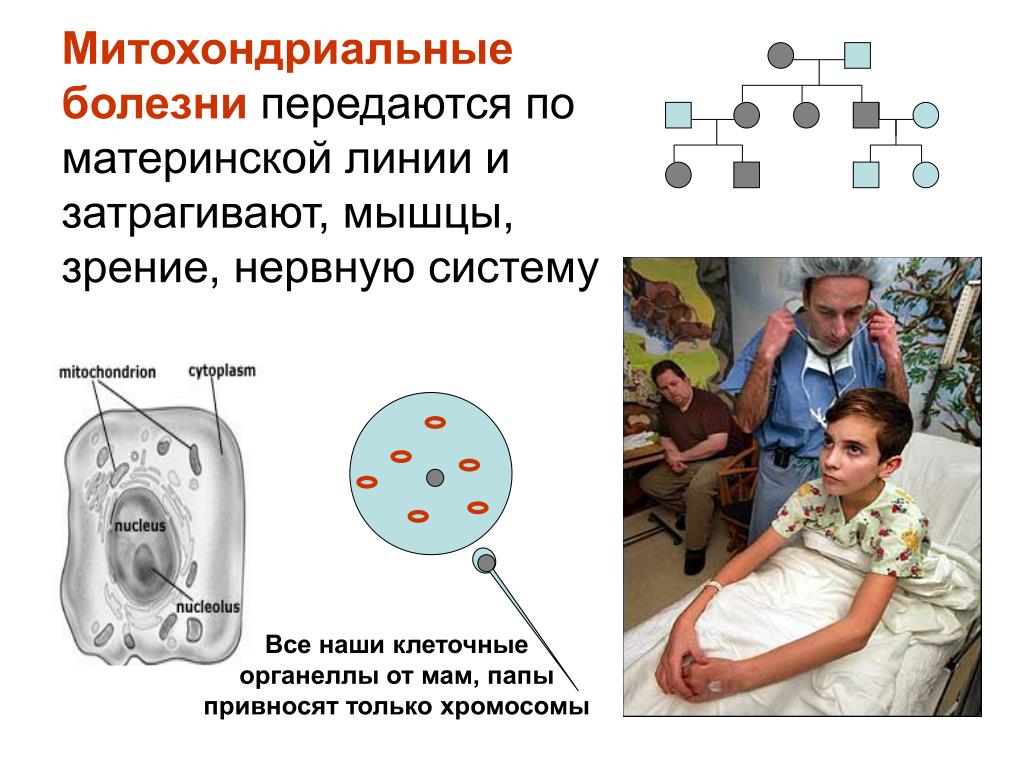 [В паблике: 9326946]
[В паблике: 9326946]
Scaglia F, Wong LJ, Vladutiu GD, Hunter JV. Преимущественная потеря объема мозжечка как нейрорадиологическая особенность дефектов дыхательной цепи у детей. AJNR Am J Нейрорадиол. 2005; 26:1675–80. [Статья бесплатно PMC: PMC7975170] [PubMed: 16091512]
Schon EA, Bonilla E, DiMauro S. Мутации митохондриальной ДНК и патогенез. J Биоэнергетическая биомембрана. 1997; 29: 131–49. [PubMed: 9239539]
Тиранти В., Вискоми С., Хильдебрандт Т., Ди Мео I, Минери Р., Тиверон С., Левитт М.Д., Прелле А., Фагиолари Г., Римольди М., Зевиани М. Потеря ETHE1, митохондриальной диоксигеназы , вызывает смертельную сульфидную токсичность при этилмалоновой энцефалопатии. Нат Мед. 2009 г.;15:200–5. [PubMed: 19136963]
Uziel G, Moroni I, Lamantea E, Fratta GM, Ciceri E, Carrara F, Zeviani M. Митохондриальное заболевание, связанное с мутацией T8993G митохондриального гена АТФазы 6: клиническое, биохимическое и молекулярное исследование в шести семьях.