
Митохондриальная энцефалопатия что это такое: MELAS-синдром как необычная причина гипопаратиреоза: клиническое наблюдение | Умярова

Митохондриальная энцефалопатия
См. фотогалерею статьи
Пациентка Г., 34 лет, впервые госпитализирована в 1невр. отд. в сентябре 2010. Жалобы при поступлении на судорожные генерализованные приступы в вечернее и ночное время, эпизоды потери сознания днём, на мышечные подёргивания лица, судороги в ногах, снижение толерантности к физическим нагрузкам, снижение памяти, нарушение навыков чтения и письма, нарушение речи. Анамнез заболевания: С 13 лет у пациентки появились приступы с потерей сознания и клонико-тоническими судорогами, через несколько месяцев присоединились миоклонические судороги с вовлечением мышц рук, ног, лица, которые были расценены как эпилепсия. На фоне приёма противосудорожных препаратов генерализованные приступы стали редкими, а миоклонии более частыми и генерализованными. В возрасте 16 и 17 лет развиваются приступы с нарушением глотания, обильным слюнотечением, нарушением речи и выраженной мышечной слабостью. Со слов матери при МРТ головного мозга выявлены очаги демиелинизации, был поставлен диагноз — рассеянный энцефаломиелит, проводились курсы плазмофереза и гормонотерапии, без эффекта. Пациентка лечилась в США, где клинико-морфологическая картина также была расценена как демиелинизирующий процесс. В 20 лет развился пароксизм с выраженными длительными и генерализующимися миоклониями. Проведено генетическое обследование, биопсия бедренной мышцы, при которой выявлены: Атрофия отдельных мышечных волокон, преимущественно 11 типа, изменение нервно-мышечных пластинок с исчезновением аксонального их компонента, при реакции на SDH только в единичных мышечных волокнах выявляется периферическая зона усиления реакции, свидетельствующая о скоплении митохондрий по периферии мышечного волокна, выявлены признаки миопатических изменений в виде нарушения архитектуры и очагового лизиса миофибриллярного аппарата, в части волокон — признаки метаболических нарушений: избыточное накопление гликогена и липидов, дегенеративные изменения митохондрий только в единичных волокнах, обнаружение скопления большого числа мелких темных митохондрий или появление митохондрий неправильной формы с включением гликогена, таким образом, в мышечной ткани преобладают неспецифические миопатические изменения, морфологические признаки митохондриальной миопатии выявлены только в единичных волокнах.
Пациентка лечилась в США, где клинико-морфологическая картина также была расценена как демиелинизирующий процесс. В 20 лет развился пароксизм с выраженными длительными и генерализующимися миоклониями. Проведено генетическое обследование, биопсия бедренной мышцы, при которой выявлены: Атрофия отдельных мышечных волокон, преимущественно 11 типа, изменение нервно-мышечных пластинок с исчезновением аксонального их компонента, при реакции на SDH только в единичных мышечных волокнах выявляется периферическая зона усиления реакции, свидетельствующая о скоплении митохондрий по периферии мышечного волокна, выявлены признаки миопатических изменений в виде нарушения архитектуры и очагового лизиса миофибриллярного аппарата, в части волокон — признаки метаболических нарушений: избыточное накопление гликогена и липидов, дегенеративные изменения митохондрий только в единичных волокнах, обнаружение скопления большого числа мелких темных митохондрий или появление митохондрий неправильной формы с включением гликогена, таким образом, в мышечной ткани преобладают неспецифические миопатические изменения, морфологические признаки митохондриальной миопатии выявлены только в единичных волокнах. Установлен диагноз: «Митохондриальная энцефалопатия. Синдром MERRF». Родители здоровы, генетически не обследованы. Периодически проходит курсы метаболической и антиоксидантной терапии. Постоянно принимает Коэнзим Q10, Депакин хроно 1500 мг в сут, Фенобарбитал 100 мг на ночь, периодически (во время приступов) Диазепам 5 мг. В течение последней недели появились миоклонические подёргивания мимической мускулатуры справа, речь стала более смазанной. Госпитализирована в экстренном порядке для обследования и лечения. Состояние при поступлении: Общее состояние относительно удовлетворительное. Кожные покровы чистые, бледные. Отеков нет. Над легкими везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. АД 110/70 мм рт. ст. Пульс 68 в мин. Живот при пальпации мягкий, безболезненный во всех отделах. Дизурии нет. Неврологический статус: Сознание ясное, ориентирована полностью. Мнестико-интелектуально снижена. Демонстративна. Речь тихая слабомодулированная. Снижение долговременной памяти.
Установлен диагноз: «Митохондриальная энцефалопатия. Синдром MERRF». Родители здоровы, генетически не обследованы. Периодически проходит курсы метаболической и антиоксидантной терапии. Постоянно принимает Коэнзим Q10, Депакин хроно 1500 мг в сут, Фенобарбитал 100 мг на ночь, периодически (во время приступов) Диазепам 5 мг. В течение последней недели появились миоклонические подёргивания мимической мускулатуры справа, речь стала более смазанной. Госпитализирована в экстренном порядке для обследования и лечения. Состояние при поступлении: Общее состояние относительно удовлетворительное. Кожные покровы чистые, бледные. Отеков нет. Над легкими везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. АД 110/70 мм рт. ст. Пульс 68 в мин. Живот при пальпации мягкий, безболезненный во всех отделах. Дизурии нет. Неврологический статус: Сознание ясное, ориентирована полностью. Мнестико-интелектуально снижена. Демонстративна. Речь тихая слабомодулированная. Снижение долговременной памяти. Менингеальных знаков нет. Поля зрения ориентировочно не сужены. Зрачки D=S. Фотореакции живые, конвергенция ослаблена. Движения глазных яблок не ограничены. Нистагм установочный мелкоразмашистый в крайних отведениях. Миоклонические подёргивания мимической мускулатуры справа. Глоточные рефлексы средней живости. Язык по средней линии. Парезов нет. Гипотония мышц конечностей. Диффузная мышечная гипотрофия. Сухожильные рефлексы живые, S=D. Патологические рефлексы не вызываются. Расстройств чувствительности нет. В позе Ромберга устойчива. Координаторные пробы выполняет удовлетворительно с 2 сторон. Дополнительное обследование: В общем и биохимическом анализе крови без патологических изменений. За исключением Лактата — 3,0. ЭКГ: Ритм синусовый. ЧСС 63 в мин. Отклонение ЭОС вправо. ЭЭГ: Диффузные изименения ЭЭГ, умеренно выраженная дизритмия, за счет неравномерности и недостаточной выраженности основного ритма покоя в сочетании с негрубым замедлением биопотенциалов мозга. В ходе записи регистрируется негрубая неспецифическая островолновая активность с преобладанием над лобно-височными отделами справа, несколько нарастающая на фоне гипервентиляции.
Менингеальных знаков нет. Поля зрения ориентировочно не сужены. Зрачки D=S. Фотореакции живые, конвергенция ослаблена. Движения глазных яблок не ограничены. Нистагм установочный мелкоразмашистый в крайних отведениях. Миоклонические подёргивания мимической мускулатуры справа. Глоточные рефлексы средней живости. Язык по средней линии. Парезов нет. Гипотония мышц конечностей. Диффузная мышечная гипотрофия. Сухожильные рефлексы живые, S=D. Патологические рефлексы не вызываются. Расстройств чувствительности нет. В позе Ромберга устойчива. Координаторные пробы выполняет удовлетворительно с 2 сторон. Дополнительное обследование: В общем и биохимическом анализе крови без патологических изменений. За исключением Лактата — 3,0. ЭКГ: Ритм синусовый. ЧСС 63 в мин. Отклонение ЭОС вправо. ЭЭГ: Диффузные изименения ЭЭГ, умеренно выраженная дизритмия, за счет неравномерности и недостаточной выраженности основного ритма покоя в сочетании с негрубым замедлением биопотенциалов мозга. В ходе записи регистрируется негрубая неспецифическая островолновая активность с преобладанием над лобно-височными отделами справа, несколько нарастающая на фоне гипервентиляции. Типичной эпиактивности на момент исследования не зафиксировано. МРТ головного мозга: На Т1 и Т2 взвешенных изображениях в сагиттальной и аксиаль¬ной проекциях исследования кора и белое вещество головного мозга развиты правильно и имеют нормальную интенсивность мр-сигнала. В базальных ядрах, внутренней капсуле и мозолистом теле не определя¬ется изменений мр-сигнала. Очаговых изменений мр сигнала в стволе и мозжечке не выявлено. Срединные структуры головного мозга не смещены. Желудочки мозга расширены, форма их не изменена. Боковые желудочки симметричны. Выявляется расширение субарахноидального пространства и бо¬розд большого мозга и мозжечка. Турецкое седло и гипофиз не измене¬ны. Параселлярные структуры без особенностей. Дополнительных образований в области мостомозжечковых уг¬лов не выявлено. Внутренние слуховые проходы не расширены, сим¬метричны. Околоносовые пазухи и ячейки сосцевидных отростков височных костей развиты правильно, их пневматизация не нарушена. Глазницы без особенностей.
Типичной эпиактивности на момент исследования не зафиксировано. МРТ головного мозга: На Т1 и Т2 взвешенных изображениях в сагиттальной и аксиаль¬ной проекциях исследования кора и белое вещество головного мозга развиты правильно и имеют нормальную интенсивность мр-сигнала. В базальных ядрах, внутренней капсуле и мозолистом теле не определя¬ется изменений мр-сигнала. Очаговых изменений мр сигнала в стволе и мозжечке не выявлено. Срединные структуры головного мозга не смещены. Желудочки мозга расширены, форма их не изменена. Боковые желудочки симметричны. Выявляется расширение субарахноидального пространства и бо¬розд большого мозга и мозжечка. Турецкое седло и гипофиз не измене¬ны. Параселлярные структуры без особенностей. Дополнительных образований в области мостомозжечковых уг¬лов не выявлено. Внутренние слуховые проходы не расширены, сим¬метричны. Околоносовые пазухи и ячейки сосцевидных отростков височных костей развиты правильно, их пневматизация не нарушена. Глазницы без особенностей. Обращает на себя внимание выраженный гиперостоз лобной и теменных костей. Заключение: Смешанная гидроцефалия. Таким образом у пациентки на 7 год проявления заболевания диагностирована – Митохондриальная энцефалопатия. Синдром MERRF. Данное заболевание диагностируется крайне редко, что вероятнее всего связано с малой информированностью неврологов и трудностями диагностики (генетическое исследование, биопсия мышц, анализ крови на лактат-пируват). Существуют разработанные профессором Научного центра неврологии РАМН С. Н. Иллариошкиным рекомендации по алгоритму диагностики митохондриальных энцефаломиопатий (см. информацию на сайте).
Обращает на себя внимание выраженный гиперостоз лобной и теменных костей. Заключение: Смешанная гидроцефалия. Таким образом у пациентки на 7 год проявления заболевания диагностирована – Митохондриальная энцефалопатия. Синдром MERRF. Данное заболевание диагностируется крайне редко, что вероятнее всего связано с малой информированностью неврологов и трудностями диагностики (генетическое исследование, биопсия мышц, анализ крови на лактат-пируват). Существуют разработанные профессором Научного центра неврологии РАМН С. Н. Иллариошкиным рекомендации по алгоритму диагностики митохондриальных энцефаломиопатий (см. информацию на сайте).
Фотогалерея статьи
4 мая 2016 г.
Подписывайтесь на наш Telegram канал!
Митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными…
МЕЛАС (митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами, англ. MELAS) — это заболевание входящее в группу митохондриальных болезней. Митохондрии это органеллы имеющие собственную ДНК, которая передается и наследуется по материнской линии. Типичные изменения при визуализации включают инсультоподобные фокусы на различных стадиях развития («shifting spread» pattern), несовпадающие с границами артериальных сосудистых бассейнов, имеется определенная предрасположенность к возникновению поражений в задних отделах теменных и затылочных долей. МР-спектроскопия может демонстрировать повышенный пик лактата даже при неизмененной картине головного мозга.
Митохондрии это органеллы имеющие собственную ДНК, которая передается и наследуется по материнской линии. Типичные изменения при визуализации включают инсультоподобные фокусы на различных стадиях развития («shifting spread» pattern), несовпадающие с границами артериальных сосудистых бассейнов, имеется определенная предрасположенность к возникновению поражений в задних отделах теменных и затылочных долей. МР-спектроскопия может демонстрировать повышенный пик лактата даже при неизмененной картине головного мозга.
Эпидемиология
Синдром MELAS обычно возникает в виде инсультоподобных эпизодов у пациентов моложе 40 лет при отсутствии факторов риска инсульта.
Клиническая картина
МЕЛАС обычно имеет рецидивирующе-ремиттирующее течение, с или без нарастающего дефицита. Клинические проявления характеризуются [1]:
- часто встречающиеся проявления
- инсультоподобные эпизоды
- судороги
- лактат ацидоз
- энцефалопатия
- деменция
- мышечная слабость
- глухота
Патология
Дефект в виде точечной мутации нуклеотида 3243 митохондриальной ДНК (A — G транслокация), кодирующего тРНК лейцина, поражает дыхательную цепь (отвечающую за производство энергии). Синтез дефектного белка приводит к нарушениям в работе различных отделов дыхательной цепи, что в итоге ведет к истощению NAD+ и NADH+. Это, в свою очередь, ведет к переключению метаболизма на анаэробный гликолиз, обуславливающий накопление лактата, что делает кору более восприимчивой к гипоксии и в конечном итоге приводит к гибели нейронов [1]. Поскольку некоторое количество митохондрий передается с яйцеклеткой, только часть митохондрии содержит мутантную ДНК. От количества мутантых генов зависит тяжесть клинических проявлений [1].
Синтез дефектного белка приводит к нарушениям в работе различных отделов дыхательной цепи, что в итоге ведет к истощению NAD+ и NADH+. Это, в свою очередь, ведет к переключению метаболизма на анаэробный гликолиз, обуславливающий накопление лактата, что делает кору более восприимчивой к гипоксии и в конечном итоге приводит к гибели нейронов [1]. Поскольку некоторое количество митохондрий передается с яйцеклеткой, только часть митохондрии содержит мутантную ДНК. От количества мутантых генов зависит тяжесть клинических проявлений [1].
Диагностика
Компьютерная томография
- множественные инфаркты
- поражение нескольких сосудистых бассейнов
- могут быть как симметричные так и асимметричные
- теменно-затылочные и височно-теменные поражения встречаются чаще
- кальцификация базальных ганглий [1,2]
- более выражены у пожилых пациентов
- атрофия [2]
Магнитно-резонансная томография
- хронические инфаркты
- поражение нескольких сосудистых бассейнов
- могут быть как симметричные так и асимметричные
- теменно-затылочные и височно-теменные поражения встречаются чаще
- острые инфаркты
- набухание извилин с повышением МР сигнала на Т2 взвешенных изображениях
- возможно контрастное усиление
- поражение субкортикального белого вещества
- повышение сигнала на диффузионно-взвешенных изображениях (T2 просвечивание) с незначительным (при наличии) изменениями на ИДК: изменения при МЕЛАС характеризуются вазогенным, а не цитотоксическим отеком, как при инфаркте [3]
МР спектроскопия: возможно повышение пика лактата даже в неизмененной мозговой паренхиме и спинно-мозговой жидкости [3].
Дифференциальный диагноз
- другие митохондриальные заболевания
- миоклоническая эпилепсия с рваными мышечными волокнами
- синдром Ли (Лея)
- синдром Кернс-Сейр
- эпилептический статус
- вирусный энцефалит
- церебральный васкулит
- болезнь Крейцфелдт- Якоба
- инфаркт за счет
- эмболии
- диссекции
- мойя-мойя
- ЦАДАСИЛ: не субкортикальные фокусы
Ключевые моменты
Диагноз MELAS основывается на следующем: 1) инсультоподобные эпизоды, возникающие в возрасте до 40 лет, 2) энцефалопатия с эпилепсией и/или деменцией, 3) наличие лактат-ацидоза, повреждений красных мышечных волокон, а также дополнительных критериев, таких, как периодические головные боли и рецидивирующая рвота. Типичные изменения при визуализации включают инсультоподобные фокусы, кальцинаты базальных ганглиев и атрофию. Расположение повреждений, не совпадающее с границами артериальных сосудистых бассейнов, а также возраст и отсутствие факторов риска выступают против инсульта. При синдроме MELAS, повреждения чаще всего возникают из-за вазогенного отека, и ИДК, как правило, не снижается или менее снижен по сравнению с недавним инфарктом.
Расположение повреждений, не совпадающее с границами артериальных сосудистых бассейнов, а также возраст и отсутствие факторов риска выступают против инсульта. При синдроме MELAS, повреждения чаще всего возникают из-за вазогенного отека, и ИДК, как правило, не снижается или менее снижен по сравнению с недавним инфарктом.
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды: MedlinePlus Genetics
Описание
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) — это состояние, поражающее многие системы организма, особенно мозг и нервную систему. системы (энцефало-) и мышц (миопатия). Признаки и симптомы этого расстройства чаще всего появляются в детстве после периода нормального развития, хотя они могут начаться в любом возрасте. Ранние симптомы могут включать мышечную слабость и боль, повторяющиеся головные боли, потерю аппетита, рвоту и судороги. У большинства больных наблюдаются эпизоды, подобные инсульту, которые начинаются до 40 лет. Эти эпизоды часто включают временную мышечную слабость на одной стороне тела (гемипарез), изменение сознания, нарушения зрения, судороги и сильные головные боли, напоминающие мигрень. Повторяющиеся эпизоды, подобные инсульту, могут постепенно повреждать мозг, приводя к потере зрения, проблемам с движением и потере интеллектуальной функции (слабоумие).
Эти эпизоды часто включают временную мышечную слабость на одной стороне тела (гемипарез), изменение сознания, нарушения зрения, судороги и сильные головные боли, напоминающие мигрень. Повторяющиеся эпизоды, подобные инсульту, могут постепенно повреждать мозг, приводя к потере зрения, проблемам с движением и потере интеллектуальной функции (слабоумие).
У большинства людей с MELAS в организме накапливается молочная кислота, что называется лактоацидозом. Повышенная кислотность крови может привести к рвоте, болям в животе, крайней усталости (усталости), мышечной слабости и затрудненному дыханию. Реже люди с MELAS могут испытывать непроизвольные мышечные спазмы (миоклонус), нарушение координации мышц (атаксия), потерю слуха, проблемы с сердцем и почками, диабет и гормональный дисбаланс.
Частота
Точная заболеваемость MELAS неизвестна. Это одно из наиболее распространенных состояний в группе, известной как митохондриальные заболевания. В совокупности митохондриальные заболевания встречаются примерно у 1 из 4000 человек.
Причины
MELAS может быть результатом мутации в одном из нескольких генов, включая MT-ND1 , MT-ND5 , MT-TH , MT-TL1 и MT1-TV 900. Эти гены находятся в ДНК клеточных структур, называемых митохондриями, которые преобразуют энергию из пищи в форму, которую могут использовать клетки. Хотя большая часть ДНК упакована в хромосомы внутри ядра, митохондрии также имеют небольшое количество собственной ДНК, известной как митохондриальная ДНК или мтДНК.
Некоторые гены, связанные с MELAS, предоставляют инструкции для создания белков, участвующих в нормальной функции митохондрий. Эти белки являются частью большого комплекса ферментов в митохондриях, которые помогают преобразовывать кислород, жиры и простые сахара в энергию. Другие гены, связанные с этим заболеванием, предоставляют инструкции для создания молекул, называемых транспортными РНК (тРНК), которые являются химическими родственниками ДНК. Эти молекулы помогают собирать белковые строительные блоки, называемые аминокислотами, в полноразмерные функционирующие белки в митохондриях.
Мутации в конкретном гене транспортной РНК, MT-TL1 , вызывают более 80 процентов всех случаев MELAS. Эти мутации нарушают способность митохондрий производить белки, использовать кислород и производить энергию. Исследователи не определили, как изменения в мтДНК приводят к конкретным признакам и симптомам MELAS. Они продолжают исследовать эффекты мутаций митохондриальных генов в различных тканях, особенно в головном мозге.
Наследование
Это состояние наследуется по митохондриальному типу, который также известен как наследование по материнской линии. Этот тип наследования относится к генам, содержащимся в мтДНК. Поскольку яйцеклетки, а не сперматозоиды, вносят митохондрии в развивающийся эмбрион, дети могут наследовать только нарушения, возникающие в результате мутаций мтДНК, от матери. Эти расстройства могут проявляться в каждом поколении семьи и поражать как мужчин, так и женщин, но отцы не передают своим детям признаки, связанные с изменениями мтДНК.
В большинстве случаев люди с MELAS наследуют измененный митохондриальный ген от своей матери. Реже заболевание возникает в результате новой мутации митохондриального гена и возникает у людей без семейного анамнеза MELAS.
Другие названия этого состояния
- MELAS
- MELAS-синдром
- Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды
- Митохондриальная миопатия, лактоацидоз, инсультоподобный эпизод
- Миопатия, митохондриальная-энцефалопатия-молочнокислый ацидоз-инсульт
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: ювенильная миопатия, энцефалопатия, лактоацидоз И инсульт
Информационный центр генетических и редких заболеваний
- Митохондриальная энцефаломиопатия лактоацидоз и инсультоподобные эпизоды
Ресурсы поддержки пациентов и защиты интересов
- Информационный поиск по болезни
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov
- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, ЭНЦЕФАЛОПАТИЯ, ЛАКТАЦИДОЗ И ИНСУЛЬТОПОДОБНЫЕ ЭПИЗОДЫ
Научные статьи в PubMed
- PubMed
Ссылки
- Беттс Дж., Ярос Э., Перри Р.Х., Шефер А.М., Тейлор Р.В., Абдель-Алл З., Лайтоулерс
РН, Тернбулл Д.М. Молекулярная нейропатология MELAS: уровень гетероплазмии в
отдельные нейроны и признаки обширного вовлечения сосудов. невропатол
Заявл. Нейробиол. 2006 авг; 32 (4): 359-73. doi: 10.1111/j.1365-2990.2006.00731.x.
Цитата в PubMed - Эль-Хаттаб А.В., Альманнаи М., Скалья Ф. МЕЛАС. 27 февраля 2001 г. [обновлено 29 ноября 2018 г.].
Пришли: Адам М.П., Эверман Д.Б., Мирзаа Г.М., Пагон Р.А., Уоллес С.Е., Бин Л.Дж.Х., Грипп К.В.,
Амемия А, редакторы. GeneReviews(R) [Интернет]. Сиэтл (Вашингтон): университет
Вашингтон, Сиэтл; 1993-2022. Доступна с
http://www.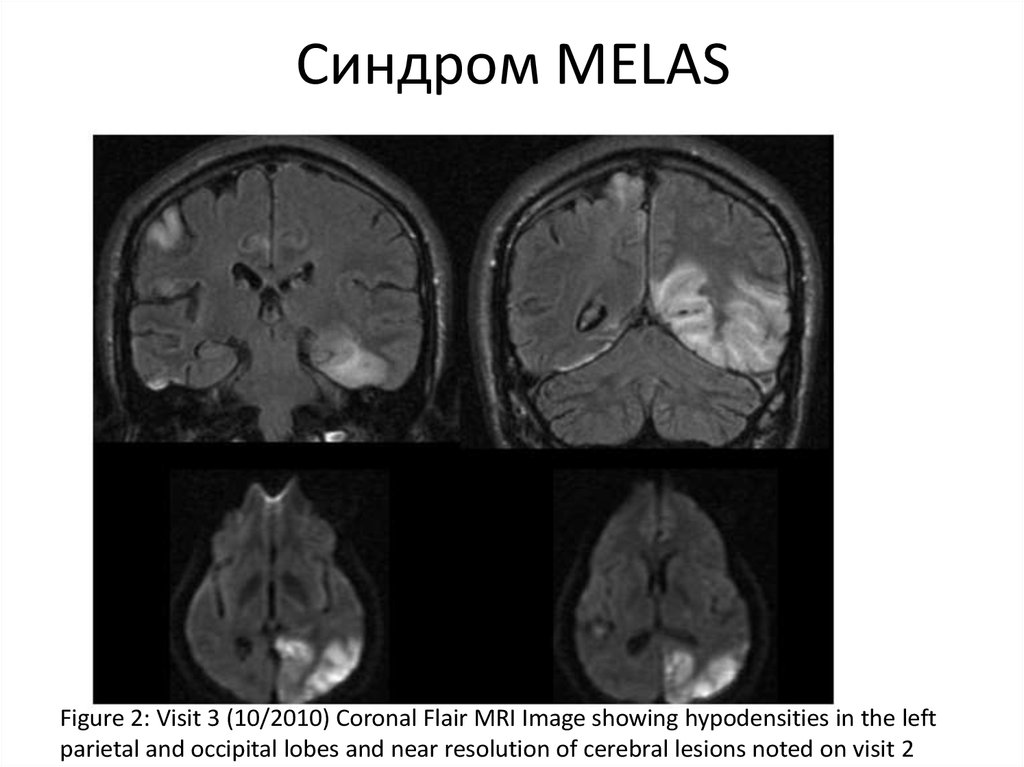 ncbi.nlm.nih.gov/books/NBK1233/
ncbi.nlm.nih.gov/books/NBK1233/
Цитата в PubMed - Гудфеллоу Дж. А., Дэни К., Стюарт В., Сантош С., Маклин Дж., Малхерн С., Разви С.
Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды:
важной причиной инсульта у молодых людей. Postgrad Med J. 2012
Июнь; 88 (1040): 326-34. doi: 10.1136/postgradmedj-2011-130326. Epub 2012, 10 февраля.
Цитата в PubMed - Мацумото Дж., Сэвер Дж.Л., Бреннан К.С., Рингман Дж.М. Митохондриальная энцефаломиопатия
с лактоацидозом и инсультом (MELAS). Преподобный Нейрол Дис. 2005 Зима; 2 (1): 30-4.
Цитата на PubMed - Sproule DM, Kaufmann P. Митохондриальная энцефалопатия, лактоацидоз и
инсультоподобные эпизоды: основные понятия, клинический фенотип и терапевтические
Лечение синдрома MELAS. Энн Н.Ю. Академия наук. 2008 Октябрь; 1142: 133-58. дои:
10.1196/летопись.1444.011. Erratum In: Ann NY Acad Sci. 2009 Апрель; 1161: 601. Цитата в PubMed - Тамбисетти М.
 , Ньюман, штат Нью-Джерси. Диагностика и лечение MELAS. Эксперт Рев Мол
, Ньюман, штат Нью-Джерси. Диагностика и лечение MELAS. Эксперт Рев Мол
Диагн. 2004 г., сен; 4(5):631-44. дои: 10.1586/14737159.4.5.631. Цитата в PubMed
Синдром MELAS — NORD (Национальная организация по редким заболеваниям)
Синдром MELAS
NORD выражает благодарность Фернандо Скалья, доктору медицинских наук, FACMG, доценту кафедры молекулярной и генетики человека Медицинского колледжа Бэйлора за помощь в подготовке настоящего документа. отчет.
Синонимы синдрома MELAS
- Митохондриальная миопатия, энцефалопатия, молочнокислый ацидоз, инсультоподобный эпизод
- Миопатия, митохондриальная энцефалопатия, молочнокислый ацидоз, инсульт
Признаки и симптомы
Причины
MELAS вызывается мутациями в митохондриальной ДНК (мтДНК). Мутации, влияющие на гены мтДНК, наследуются от матери. МтДНК, обнаруженная в сперматозоидах, обычно теряется во время оплодотворения, и в результате вся мтДНК человека происходит от матери. Больная мать передаст мутацию всем своим детям, но только ее дочери передадут мутацию своим детям. Митохондрии, которые обнаруживаются сотнями или тысячами в клетках тела, особенно в мышечной и нервной ткани, несут в себе схемы регуляции производства энергии.
Больная мать передаст мутацию всем своим детям, но только ее дочери передадут мутацию своим детям. Митохондрии, которые обнаруживаются сотнями или тысячами в клетках тела, особенно в мышечной и нервной ткани, несут в себе схемы регуляции производства энергии.
В одной и той же клетке может существовать как нормальная, так и мутированная мтДНК, что называется гетероплазмией. Количество дефектных митохондрий может превышать количество нормальных митохондрий. Симптомы могут не проявляться ни в одном поколении, пока мутация не затронет значительную часть мтДНК. Неравномерное распределение нормальной и мутантной мтДНК в разных тканях может влиять на разные органы у членов одной семьи. Это может привести к различным симптомам у пострадавших членов семьи.
Мутации в гене MT-TL1 мтДНК связаны с MELAS примерно в 80% случаев. Мутации в MT-TQ, MT-TH, MT-TK, MT-TS1, MT-ND1, MT-ND5, MT-ND6 и MT-TS2 также связаны с синдромом MELAS.
Некоторые случаи синдрома MELAS возникают в результате новой спонтанной мутации в митохондриальном гене и не передаются по наследству.
Кроме того, мутации в ядерном гене (POLG1) были связаны с синдромом MELAS в одном случае.
Пораженные группы населения
Синдром MELAS — редкое заболевание, поражающее мужчин и женщин в равной степени. Хотя синдром MELAS встречается редко, он, вероятно, является наиболее распространенным типом митохондриальной миопатии, вызванной мутациями в мтДНК. Некоторые исследователи считают, что митохондриальные миопатии могут оставаться незамеченными и не диагностируемыми среди населения в целом, что затрудняет определение истинной частоты расстройств, таких как синдром MELAS.
Диагностика
MELAS диагностируется на основании клинических данных и результатов молекулярно-генетического тестирования.
Клинические испытания могут включать измерение концентрации лактата и пирувата и белка ЦСЖ, которые повышены при синдроме MELAS. Методы визуализации головного мозга, такие как магнитно-резонансная томография (МРТ), могут использоваться для поиска поражений, подобных инсульту, а магнитно-резонансная спектроскопия (МРС) может использоваться для поиска пика лактата в мозге. Электрокардиограмма может использоваться для диагностики нарушений сердечного ритма, а эхокардиограмма может использоваться для диагностики кардиомиопатии. Биопсия мышц обычно показывает рваные красные волокна.
Электрокардиограмма может использоваться для диагностики нарушений сердечного ритма, а эхокардиограмма может использоваться для диагностики кардиомиопатии. Биопсия мышц обычно показывает рваные красные волокна.
Мутации мтДНК, связанные с MELAS, обычно можно обнаружить в лейкоцитах, но из-за гетероплазмии (см. Причины) могут потребоваться образцы других тканей, таких как кожа, волосяные фолликулы, мочевой осадок и скелетные мышцы. Осадок мочи лучше всего подходит для обнаружения мутации по сравнению с кровью, кожей и волосяными фолликулами.
Стандартная терапия
Лечение
Специального лечения синдрома MELAS не существует. Противосудорожные препараты используются для предотвращения и контроля судорог, связанных с синдромом MELAS. Вальпроевую кислоту не следует использовать в качестве противосудорожного средства. Кохлеарные имплантаты используются для лечения нейросенсорной глухоты. Терапия иногда используется для увеличения выработки энергии митохондриями и замедления последствий состояния. Коэнзим Q10 и L-карнитин оказались полезными у некоторых пациентов. У пациентов с митохондриальными миопатиями в целом умеренная тренировка на беговой дорожке может привести к улучшению аэробной способности и снижению уровня лактата в покое. Сообщалось, что внутривенное введение L-аргинина улучшает симптомы заболевания во время острых эпизодов, подобных инсульту. Сообщалось, что пероральный прием аргинина снижает частоту повторения инсультоподобных эпизодов при применении в бессимптомный период.
Коэнзим Q10 и L-карнитин оказались полезными у некоторых пациентов. У пациентов с митохондриальными миопатиями в целом умеренная тренировка на беговой дорожке может привести к улучшению аэробной способности и снижению уровня лактата в покое. Сообщалось, что внутривенное введение L-аргинина улучшает симптомы заболевания во время острых эпизодов, подобных инсульту. Сообщалось, что пероральный прием аргинина снижает частоту повторения инсультоподобных эпизодов при применении в бессимптомный период.
Генетическое консультирование рекомендуется больным людям и их семьям.
Исследовательская терапия
Лекарства, включая карнитин, коэнзим Q10, менадион, аскорбиновую кислоту, рибофлавин, тамин, никотинамид, моногидрат креатина, идебенон, сукцинат и дихлорацетат, оказались полезными у отдельных пациентов, но необходимы дальнейшие исследования для подтверждения их эффективности. Аргинин и цитруллин исследуются как потенциальные средства для уменьшения повреждения головного мозга при инсультоподобных эпизодах.
Колумбийский университет в Нью-Йорке ищет участников для двойного слепого плацебо-контролируемого клинического исследования идебенона при MELAS (митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды). В исследовании фазы IIa будут сравниваться две разные дозы экспериментального препарата идебенона, вводимые в течение одного месяца, чтобы определить эффективность препарата. Люди с MELAS и мутацией 3243 в возрасте от 8 до 65 лет могут иметь право на участие. Основная цель клинического испытания — определить, влияет ли идебенон на лактат головного мозга, измеренный с помощью магнитно-резонансной спектроскопии (МРС). МРС проводится с помощью МРТ-сканера, она безопасна и обычно хорошо переносится. Дополнительной целью является изучение безопасности и переносимости идебенона у людей с MELAS. Главным исследователем является доктор Митио Хирано. Если вам или члену вашей семьи нужна дополнительная информация об этом исследовании, посетите наш веб-сайт или свяжитесь с координатором исследований Крисом Энгельстадом по номеру, указанному ниже.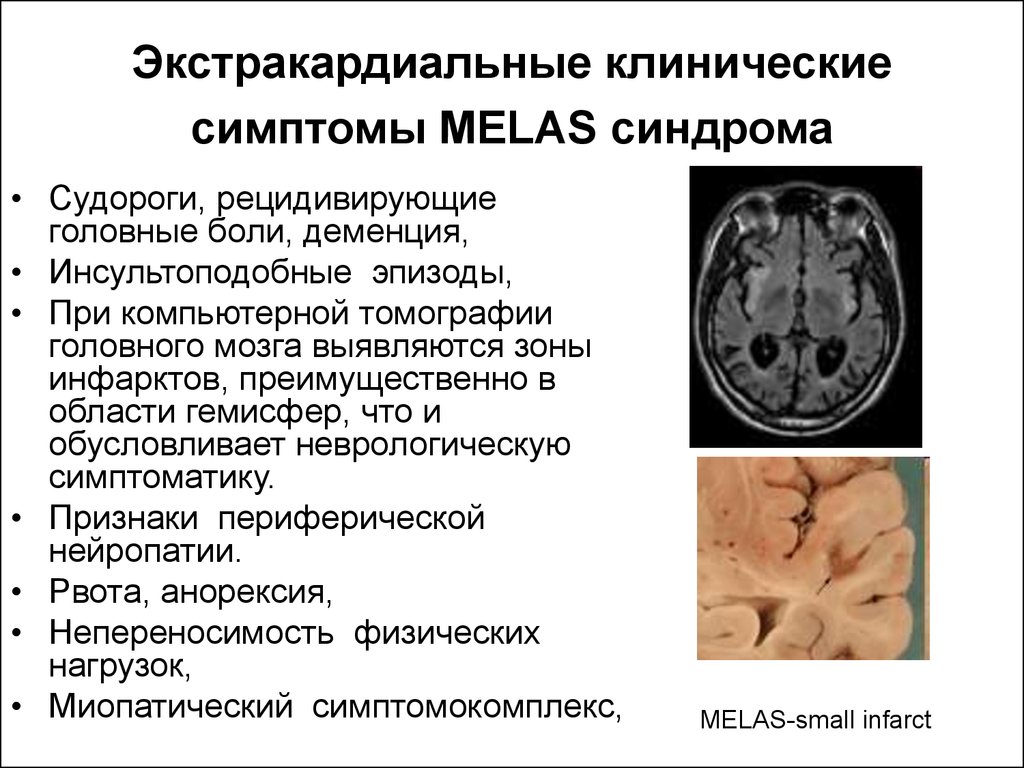
Контактное лицо: Крис Энгельстад, координатор исследований
(212) 305-6834
Медицинский колледж Бейлора и Детская больница Техаса набирают людей с синдромом MELAS для двух клинических исследований. Первое исследование направлено на измерение уровня оксида азота у пострадавших людей и определение того, увеличит ли введение аргинина или цитруллина образование оксида азота. Считается, что оксид азота полезен для улучшения и предотвращения инсультов, поэтому, если показано, что аргинин и/или цитруллин увеличивают образование оксида азота, их можно использовать для предотвращения и улучшения состояния при инсультах у пациентов с синдромом MELAS. Диабет часто встречается при синдроме MELAS, и целью второго исследования является оценка того, как больные люди расщепляют и накапливают сахар в своем организме. Это может привести к лучшему пониманию причин диабета в MELAS, что может повлиять на прогноз и лечение диабета у субъектов с MEAS. В исследовании могут участвовать взрослые и дети, страдающие синдромом MELAS и несущие мутацию m. 3243 A>G. Участники исследования будут госпитализированы в Общий клинический исследовательский центр (GCRC) в Детской больнице Техаса, где уровень оксида азота и уровень глюкозы будут измерять с помощью инфузии стабильных изотопов, что является безопасной процедурой. Главный исследователь — доктор Фернандо Скалья. Для получения дополнительной информации обращайтесь:
3243 A>G. Участники исследования будут госпитализированы в Общий клинический исследовательский центр (GCRC) в Детской больнице Техаса, где уровень оксида азота и уровень глюкозы будут измерять с помощью инфузии стабильных изотопов, что является безопасной процедурой. Главный исследователь — доктор Фернандо Скалья. Для получения дополнительной информации обращайтесь:
Доктор Айман Эль-Хаттаб
[email protected]
Телефон: 832-822-4289
Страница: 832-824-7243(5523)
Информация о текущих клинических исследованиях размещена в Интернете по адресу www. Clinictrials.gov. Все исследования, финансируемые правительством США, а некоторые из них поддерживаются частным сектором, публикуются на этом правительственном веб-сайте.
Для получения информации о клинических испытаниях, проводимых в Клиническом центре NIH в Бетесде, штат Мэриленд, обращайтесь в отдел набора пациентов NIH:
Бесплатный звонок: (800) 411-1222
Телетайп: (866) 411-1010
Эл.
Контакт для получения дополнительной информации о синдроме MELAS:
Fernando Scaglia, MD, FACMG
Доцент
Факультет молекулярной и генетики человека
Медицинский колледж Бейлора
адрес электронной почты: [email protected]
Ссылки
УЧЕБНИКИ
Beers MH, Berkow R, eds. Руководство Merck, 17-е изд. Станция Уайтхаус, Нью-Джерси: исследовательские лаборатории Merck; 1999:2478.
Fauci AS, et al, ред. Принципы внутренней медицины Харрисона, 14-е изд. Нью-Йорк, штат Нью-Йорк: McGraw-Hill, Inc.; 1998:2454, 2480.
Adams, RD, et al, ред. Принципы неврологии. 6-е изд. Нью-Йорк, штат Нью-Йорк: McGraw-Hill, компании; 1997:986.
Bennett JC, Plum F, ред. Сесил Учебник медицины. 20-е изд. Филадельфия, Пенсильвания: WB. Сондерс Ко; 1996:2167.
Берман RE, изд. Учебник педиатрии Нельсона, 15-е изд. Филадельфия, Пенсильвания: WB. Компания Сондерс; 1996:1715, 1754.
Lyon G, et al, ред. Неврология наследственных болезней обмена веществ в детском возрасте. 2-е изд. Нью-Йорк, штат Нью-Йорк: компании McGraw-Hill; 1996: 256.
2-е изд. Нью-Йорк, штат Нью-Йорк: компании McGraw-Hill; 1996: 256.
Menkes JH, au, Pine JW, et al, eds. Учебник детской неврологии, 5-е изд. Балтимор, Мэриленд: Williams & Wilkins; 1995:852-853.
Scriver CR, et al, ред. Метаболические и молекулярные основы наследственных заболеваний. 7-е изд. Нью-Йорк, штат Нью-Йорк; Макгроу-Хилл Компани, Инк; 1995:1562-1564
Buyse ML, изд. Энциклопедия врожденных дефектов. Довер, Массачусетс: Научные публикации Блэквелла; Для: Центр информационных услуг по врожденным дефектам Inc; 1990:1195.
СТАТЬИ В ЖУРНАЛЕ
Deschauer M, Tennant S, Rokicka A, et al. MELAS связан с мутациями в гене POLG1. Неврология. 2007;68(20):1741-2.
Scaglia F, Northrop JL. Митохондриальная миопатическая энцефалопатия, лактоацидоз с синдромом инсультоподобных эпизодов (MELAS): обзор вариантов лечения. Препараты ЦНС. 2006;20(6):443-64.
Кога Ю., Акита Ю., Нишиока Дж. и др. L-аргинин улучшает симптомы инсультоподобных эпизодов MELAS. Неврология. 2005;64(4):710-2.
Неврология. 2005;64(4):710-2.
Сью К.М. и др. Детская энцефалопатия, связанная с мутацией MELAS A3243G. J Педиатр. 1999; 134:696-700.
Сингх С.К. и др. синдром МЕЛАС. Индийский J Педиатр. 1999;66:621-625.
Deschauer M, et al. Митохондриальная мутация 3243 AG (мутация MELAS), связанная с болезненной ригидностью мышц. Нервно-мышечное расстройство. 1999;9:305-307.
Абэ К. и др. Эффект коэнзима Q10 у пациентов с митохондриальной миопатией, энцефалопатией, лактоацидозом и инсультоподобными эпизодами (MELAS): оценка с помощью неинвазивной тканевой оксиметрии. J Neurol Sci. 1999;162:65-68.
Хауэлл Н., Митохондриальные заболевания человека: ответы на вопросы и ответы на вопросы. Int Rev Cytol. 1999;186:49-116.
Сайтох С. и др. Эффекты дихлорацетата у трех пациентов с MELAS. Неврология. 1998:50:531-534.
Сперл В., Диагностика и лечение митохондриопатий. Вена Клин Wochenschr. 1997;109:93-99.
Чиафалони Э. и др. MELAS: клинические особенности, биохимия и молекулярная генетика. Энн Нейрол. 1992; 31:391-398.
Энн Нейрол. 1992; 31:391-398.
Гото Ю. и др. Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды (MELAS): корреляционное исследование клинических признаков и мутации митохондриальной ДНК. Неврология. 1992;42:545-550.
Хирано М. и др. MELAS: оригинальный случай и клинические критерии диагноза. Нервно-мышечное расстройство. 1992;2:125-135.
Палца Дж. Другой геном человека. Наука. 1990;249:1104-1105.
Дрисколл П.Ф. и др. Синдром MELAS у матери и двоих детей. Арх Нейрол. 1987; 44:971-973.
ИЗ ИНТЕРНЕТА
Скалья Ф. Синдром Меласа. Журнал электронной медицины. Последнее обновление: 03.05.10: Доступно по адресу http://emedicine.medscape.com/article/946864-overview По состоянию на 10 сентября.
МакКьюсик В.А., изд. Онлайн менделевское наследование у человека (OMIM). Балтимор. Доктор медицины: Университет Джона Хопкинса; Запись №:540000; Последнее обновление: 13.04.10.
Genetics Home Reference-U.S. База данных библиотеки медицины. http://ghr.nlm.nih.gov/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes. Обновлено 11/06. Доступно 9/10.
http://ghr.nlm.nih.gov/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes. Обновлено 11/06. Доступно 9/10.
ДиМауро С. и Хирано М. (обновлено 14.10.10). МЕЛАС. В GeneReviews at Genetests: Информационный ресурс по медицинской генетике (база данных онлайн). Авторское право, Вашингтонский университет, Сиэтл. 1997-2010 гг. Доступно на http://www.genetests.org. Доступ 11/10.
Годы публикации
1993, 1998, 1999, 2000, 2001, 2011
Информация в базе данных редких заболеваний NORD предназначена только для образовательных целей и не предназначена для замены рекомендаций врача или другого квалифицированного медицинского работника.
Содержание веб-сайта и баз данных Национальной организации редких заболеваний (NORD) защищено авторским правом и не может быть воспроизведено, скопировано, загружено или распространено каким-либо образом в коммерческих или общественных целях без предварительного письменного разрешения и одобрения.