
Миокардиопатии у детей: причины, симптомы лечение в Москве

Кардиомиопатии при врожденных нарушениях метаболизма у детей | Леонтьева
1. Maron B.J., Towbin J.A., ThieneG. et al. Contemporary definitions and classification of the cardiomyopathies. An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816.
2. Elliott P., Andersson B., Arbustini E. et al Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29: 270–276.
3. Wilkinson J.D., Landy D.C., Colan S.D. et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin 2010; 6: 401–441.
4. Towbin J.A., Lowe A.M., Colan S.D., Sleeper L. A. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006; 296: 15: 1867–1876.
A. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006; 296: 15: 1867–1876.
5. Lipshultz S.E., Orav E.J., Wilkinson J.D. et al. Risk stratification at the time of diagnosis for children with hypertrophic cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry Study Group. Lancet 2013; 382: 9908: 1889–1897.
6. Dipchand A.I., Naftel D.C., Feingold B. et al. Outcomes of children with cardiomyopathy listed for transplant: a multi-institutional study. J Heart Lung Transplant 2009; 28: 1312–1321.
7. Bharucha T., Lee K.J., Daubeney P.E. et al. Sudden death in childhood cardiomyopathy: results from a long-term national population-based study. J Am Coll Cardiol 2015; 65: 21: 2302–2301.
8. Byers S.L., Ficicioglu C. Infant with cardiomyopathy: When to suspect inborn errors of metabolism? World J Cardiol 2014; 6: 11: 1149–1155.
9. Cox G.F. Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol 2007; 24: 15–25.
Prog Pediatr Cardiol 2007; 24: 15–25.
10. Kindel S.J., Miller E.M., Gupta R. et al. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail 2012; 18: 396–403.
11. Леонтьева И.В., Белозеров Ю.М., Сухоруков В.С., Николаева Е.А. Проблемы современной диагностики метаболических кардиомиопатий. Рос вестн перинатол и педиатр 2012; 4(1): 55–63. (Leont’eva I.V., Belozerov Yu.M., Sukhorukov V.S., Nikolaeva E.А. Problem of current diagnosis of metabolic cardiomyopathies. Ros vestn perinatol i pediatr 2012; 4(1): 55–63).
12. Wicks E.C., Elliott P.M. Genetics and metabolic cardiomyopathies. Herz 2012; 37: 598–610.
13. Meyers D.E., Basha H.I., Koenig M.K. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J 2013; 40: 385–394.
14. Colan S.D., Lipshultz S.E., Lowe A.M. et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation 2007; 115: 6: 773–781.
Circulation 2007; 115: 6: 773–781.
15. Bonnet D., Martin D., de Lonlay P. et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 1999; 100: 2248–2253.
16. Sharma S., Black S.M. Сarnitine homeostasis, mitochondrial function, and cardiovascular disease. Drug Discov Today Dis Mech 2009; 6: 1-4: e31–e39.
17. Löster H. Carnitine and cardiovascular diseases. Ponte Press Verlags-GmbH, 2003; 336.
18. Cederbaum S.D., Koo-McCoy S., Tein I. et al. Carnitine membrane transporter deficiency: a long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol Genet Metab 2002; 77: 195–201.
19. Pierpont M.E., Breningstall G.N., Stanley C.A. A Familial carnitine transporter defect: A treatable cause of cardiomyopathy in children. Am Heart J 2000; 139: 2: Pt 3: S96–106.
20. Bautsta J., Rafel E., Martines A. Famial hypertrophic cardiomyopathy and muscle carnitine deficiency. Muscle Nerve 1990; 13: 192–194.
Muscle Nerve 1990; 13: 192–194.
21. Angelini C., Vergani L., Martinuzzi A. et al. Clinical and biochemical aspects of carnitine deficiency and insufficiency: transport defects and inborn errors of beta-oxidation. Crit Rev Clin Lab Sci 1992; 29: 3-4: 217–242.
22. Gesuete V., Ragni L., Picchio F.M. The “big heart” of carnitine. G Ital Cardiol (Rome) 2010; 11: 9: 703–705.
23. Николаева Е.А., Леонтьева И.В., Калачанова Е.П., Золкина И.В. Задержка физического развития и кардиомиопатия у ребенка с первичным системным дефицитом карнитина. Трудный пациент 2012; 2-3: 50–54. (Nikolaeva E.А., Leont’eva I.V., Kalachanova E.P., Zolkina I.V. Retardation of physical development and cardiomyopathy in child with primery systemic carnitine deficiency. Trudnyj patsient 2012; 2-3: 50–54.)
24. Леонтьева И.В., Алимина Е.Г., Николаева Е.А. и др. Клиническое значение нарушений метаболизма карнитина в развитии кардиомиопатий у детей. Рос вестн перинатол и педиатр 2013; 1: 34–40. (Leont’eva I. V., Аlimina E.G., Nikolaeva E.А. et al. Clinical significance of carnitine metabolism disturbances in cardiomyopathies formation at children. Ros vestn perinatol i pediatr 2013; 1: 34–40.)
V., Аlimina E.G., Nikolaeva E.А. et al. Clinical significance of carnitine metabolism disturbances in cardiomyopathies formation at children. Ros vestn perinatol i pediatr 2013; 1: 34–40.)
25. Schulze A., Lindner M., Kohlmuller D. et al. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome and implications. Pediatrics 2003; 111: 6: 1399–1406.
26. Sharma R., Perszyk A., Marangi D. et al. Lethal neonatal carnitine palmitoyltransferase II deficiency: an unusual presentation of a rare disorder. Am J Perinatol 2003; 20: 25–32.
27. Bonnefont J.P., Djouadi F., Prip-Buus C. et al. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004; 25: 495–520.
28. Rubio-Gozalbo M.E., Bakker J.A., Waterham H.R., Wanders R.J. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Aspects Med 2004; 25: 5–6: 521–532.
29. Николаева Е.А., Мамедов И.С. Диагностика наследственных дефектов обмена жирных кислот у детей. Рос вестн перинатол и педиатр 2008; 6: 37–40. (Nikolaeva E.А., Mamedov I.S. Diagnostics of fatty acids metabolic defects at children. Ros vestn perinatol i pediatr 2008; 6: 37–40.)
Николаева Е.А., Мамедов И.С. Диагностика наследственных дефектов обмена жирных кислот у детей. Рос вестн перинатол и педиатр 2008; 6: 37–40. (Nikolaeva E.А., Mamedov I.S. Diagnostics of fatty acids metabolic defects at children. Ros vestn perinatol i pediatr 2008; 6: 37–40.)
30. Parini R., Vegni C., Martini J. et al. Sudden infant death and multiple acyl-CoA dehydrogenation disorders. Eur J Pediatr 1995; 154: 421–422.
31. Леонтьева И.В. Белозеров Ю.М. Диагностика и лечение метаболических кардиомиопатий у детей, возникающих на фоне нарушения обмена жирных кислот. Лечащий врач 2012; 9: 57–63. (Leont’eva I.V. Belozerov Yu.M. Diagostics and treatment of metabolic cardiomyopathies at children with fatty acids metabolism disturbances. Lechashhij vrach 2012; 9: 57–63).
32. Kompare M., Rizzo W.B. Mitochondrial fatty-acid oxidation disorders. Semin Pediatr Neurol 2008; 15: 140–149.
33. Fletcher J.M. Screening for lysosomal storage disorders: a clinical perspective. J Inherit Metab Dis 2006; 29: 2–3: 405–408.
34. Vellodi A. Lysosomal storage disorders. Br J Haematol 2005; 128: 4: 413–431.
35. Tariq M., Ware S. Importance of genetic evaluation and testing in pediatric cardiomyopathy. World J Cardiol 2014; 6: 11: 1156–1165.
36. Arad M., Maron B., Gorham J. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 2005; 352: 362–372.
37. Kishnani P.S., Steiner R.D. Pompe disease diagnosis and management guideline. Genetics in Medicine 2006; 8: 5: 267–288.
38. Басаргина Е.Н., Жарова О.П., Архипова Е.Н. и др. Опыт применения ферментозаместительной терапии рекомбинантной человеческой кислой альфа-глюкозидазой у детей с инфантильной формой болезни Помпе. Рос вестн перинатол и педиатр 2013; 58: 6: 58–66. (Basargina E.N.,
39. Zharova O.P., Аrkhipova E.N. et al. Experience of application of enzyme replacement therapy with recombinant human acid alpha-glucosidase in children with infantile form of Pompe disease. Ros vestn perinatol i pediatr 2013; 58: 6: 58–66. )
)
40. Noori S., Acherman R., Siassi B. et al. A rare presentation of Pompe disease with massive hypertrophic cardiomyopathy at birth. J Perinat Med 2002; 30: 6: 517–521.
41. Van Maldergem L., Haumont D., Saurty D. et al. Bradycardia in a case of type II glycogenosis (Pompe’s disease) revealing in early neonatal period. Acta Clin Belg 1990; 45: 6: 412–414.
42. Fung K.P., Lo R.N., Ho H.C. Pompe’s disease presenting as supraventricular tachycardia. Aust Paediatr J 1989; 25: 2: 101–102.
43. Metzl J.D., Elias E.R., Berul C.I. An interesting case of infant sudden death: severe hypertrophic cardiomyopathy in Pompe’s disease. Pacing Clin Electrophysiol 1999; 22: 5: 821–822.
44. Семячкина А.Н., Сухоруков В.С., Букина Т.М. и др. Болезнь накопления гликогена, тип II (болезнь Помпе) у детей. Рос вестн перинатол и педиатр 2014; 59: 4: 48–55. (Semyachkina А.N., Sukhorukov V.S., Bukina T.M. et al. Glycogen storage disease, type II (Pompe disease) in children. Ros vestn perinatol i pediatr 2014; 59: 4: 48–55. )
)
45. Jacob J.L., Leandro R.L., Parro Junior A. Pompe’s disease or type IIa glycogenosis. Arq Bras Cardiol 1999; 73: 5: 435–440.
46. Tabarki B., Mahdhaoui A., Yacoub M. et al. Familial hypertrophic cardiomyopathy associated with Wolff-Parkinson-White syndrome revealing type II glycogenosis. Arch Pediatr 2002; 9: 7: 697–700.
47. Hagemans M.L., Winkel L.P., Van Doorn P.A. et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005; 128: Pt 3: 671–677.
48. Klinge L., Straub V., Neudorf U., Vot T. Enzyme replacement therapy in classical infantile pompe disease: results of a tenmonth follow-up study. Neuropediatrics 2005; 36: 1: 6–11.
49. Blair E., Redwood C., Ashrafian H. et al. Mutations in the gamma (2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001; 10: 1215–1220.
50. Sugie K. , Yamamoto A., Murayama K. et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002; 58: 12: 1773–1778.
, Yamamoto A., Murayama K. et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002; 58: 12: 1773–1778.
51. Boucek D., Jirikowic J., Taylor M. Natural history of Danon disease. Genet Med 2011; 13: 6: 563–568.
52. Tanaka Y., Guhde G., Suter A. et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406: 6798: 902–906.
53. Maron B.J., Roberts W.C., Arad M. et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009; 301: 12: 1253.
54. Леонтьева И.В., Царегородцев Д.А. Болезнь Данона как причина гипертрофической кардиомиопатии. Рос вестн перинатол и педиатр 2015; 3: 36–42. (Leont’eva I.V., Tsaregorodtsev D.А. Danon disease as a cause of hypertrophic cardiomyopathy. Ros vestn perinatol i pediatr 2015; 3: 36–42.)
55. Maron B.J., Roberts W.C., Ho C.Y. et al. Profound left ventricular remodeling associated with LAMP2 cardiomyopathy. Am J Cardiol 2010; 106: 1194–1196.
56. Cheng Z., Fang Q. Danon disease: focusing on heart. J Hum Genet 2012; 57: 7: 407–410.
57. Van Der Starre P., Deuse T., Pritts C. et al. Late profound muscle weakness following heart transplantation due to Danon disease. Muscle Nerve 2013; 47: 1: 135–137.
58. Zaki A., Zaidi A., Newman W.G., Garratt C.J. Advantages of a subcutaneous implantable cardioverter-defibrillator in LAMP2 hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol 2013; 24: 9: 1051–1053.
59. Леонтьева И.В., Царегородцев Д.А. Лизосом-ассоциированная гипертрофическая кардиомиопатия у двух сибсов. Рос вестн перинатол и педиатр 2015; 4: 75–82. (Leont’eva I.V., Tsaregorodtsev D.А. Lysosome-associated hypertrophic cardiomyopathy in two sibs. Ros vestn perinatol i pediatr 2015; 4: 75–82.)
60. Lee P.J., Deanfield J.E., Burch M. et al. Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am J Cardiol 1997; 79: 6: 834–838.
61. Amin A.S., Kasturi L., Kulkarni A.V., Ajmera N.K. Glycogen storage disease type III. Indian Pediatr 2000; 37: 6: 670–673.
62. Moses S.W., Wanderman K.L., Myroz A., Frydman M. Cardiac involvement in glycogen storage disease type III. Eur J Pediatr 1989; 148: 8: 764–766.
63. Tada H., Kurita T., Ohe T. et al. Glycogen storage disease type III associated with ventricular tachycardia. Am Heart J 1995; 130: 4: 911–912.
64. Akazawa H., Kuroda T., Kim S. et al. Specific heart muscle disease associated with glycogen storage disease type III: clinical similarity to the dilated phase of hypertrophic cardiomyopathy. Eur Heart J 1997; 18: 3: 532–533.
65. Sachdev B., Takenaka T., Teraguchi H. et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002; 105: 1407–1411.
66. Ries M., Gupta S., Moore D.F. et al. Pediatric Fabry disease. Pediatrics 2005; 115: 3: e344–355.
67. Palecek T., Lubanda J. C., Magage S. et al. Cardiac manifestation of Fabry’s disease: current knowledge. Vnitr Lek 2004; 50: 11: 846–851.
C., Magage S. et al. Cardiac manifestation of Fabry’s disease: current knowledge. Vnitr Lek 2004; 50: 11: 846–851.
68. Kampmann C., Baehner F.A., Whybra C. et al. The right ventricle in Fabry disease. Acta Paediatr Suppl 2005; 94: 447: 15–18.
69. Kalliokoski R.J., Kalliokoski K.K., Sundell J. et al. Impaired myocardial perfusion reserve but preserved peripheral endothelial function in patients with Fabry disease. J Inherit Metab Dis 2005; 28: 4: 563–573.
70. Blum A., Ashkenazi H., Haromankov I. et al. First-degree atrioventricular block and restrictive physiology as cardiac manifestations of Fabry’s disease. South Med J 2003; 96: 2: 212–213.
71. Shah J.S., Elliott P.M. Fabry disease and the heart: an overview of the natural history and the effect of enzyme replacement therapy. Acta Paediatr Suppl 2005; 94: 447: 11–14.
72. Hoffmann B., Garcia de Lorenzo A., Mehta A. et al. Effects of enzyme replacement therapy on pain and health related quality of life in patients with Fabry disease: data from FOS (Fabry Outcome Survey). J Med Genet 2005; 42: 3: 247–252.
J Med Genet 2005; 42: 3: 247–252.
73. Довгань М.И., Белозеров Ю.М., Семячкина А.Н. Поражение сердца при мукополисахаридозах. Рос вестн перинатол и педиатр 2014; 59: 3: 22–31. (Dovgan’ M.I., Belozerov Yu.M., Semyachkina А.N. Heart damage in mucopolysaccharidosis. Ros vestn perinatol i pediatr 2014; 59: 3: 22–31.)
АКТУАЛЬНОСТЬ ГЕНЕТИЧЕСКОЙ ВЕРИФИКАЦИИ НЕКОМПАКТНОЙ КАРДИОМИОПАТИИ У ДЕТЕЙ: КЛИНИЧЕСКИЕ СЛУЧАИ | Сдвигова
1. Finsterer J. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol. 2009;30(5):659–681. doi: 10.1007/s00246-008-9359-0.
2. Ерохина М.Г. Некомпактный миокард левого желудочка: структурно-функциональное состояние миокарда и особенности клинических проявлений: Автореф. дис. … канд. мед. наук. — М.; 2009. — 26 с.
3. Ali SK, Abu-Sulaiman R, Agouba RB. Noncompaction cardiomyopathy: a new mechanism for mitral regurgitation with distinct clinical, echocardiographic features and pathological correlations. J Saudi Heart Assoc. 2015;27(2):71–78. doi: 10.1016/j.jsha.2014.07.002.
J Saudi Heart Assoc. 2015;27(2):71–78. doi: 10.1016/j.jsha.2014.07.002.
4. Amzulescu MS, Rousseau MF, Ahn SA, et al. Prognostic impact of hypertrabeculation and noncompaction phenotype in dilated cardiomyopathy: a CMR study. JACC Cardiovasc Imaging. 2015;8(8): 934–946. doi: 10.1016/j.jcmg.2015.04.015.
5. Alhabshan F, Smallhorn JF, Golding F, et al. Extent of myocardial non-compaction: comparison between MRI and echocardiographic evaluation. Pediatr Radiol. 2005;35(11):1147–1151. doi: 10.1007/s00247-005-1551-2.
6. Murphy RT, Thaman R, Blanes JG, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26(2):187–192. doi: 10.1093/eurheartj/ ehi025.
7. Stanton C, Bruce C, Connolly H, et al. Isolated left ventricular noncompaction syndrome. Am J Cardiol. 2009;104(8):1135–1138. doi: 10.1016/j.amjcard.2009.05.062.
8. Engberding R, Stollberger C, Ong P, et al. Isolated non-compaction cardiomyopathy. Dtsch Arztebl Int. 2010;107(12):206–213. doi: 10.3238/arztebl.2010.0206.
2010;107(12):206–213. doi: 10.3238/arztebl.2010.0206.
9. Сильнова И.В. Ультразвуковая диагностика некомпактного миокарда у детей: Автореф. дис. … канд. мед. наук. — М.; 2012. — 23 c.
10. Jefferies JL, Wilkinson JD, Sleeper LA, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the pediatric cardiomyopathy registry. J Card Fail. 2015;21(11):877–884. doi: 10.1016/j.cardfail.2015.06.381.
11. McMahon CJ, Pignatelli RH, Nagueh SF, et al. Left ventricular non-compaction cardiomyopathy in children: characterisation of clinical status using tissue Doppler-derived indices of left ventricular diastolic relaxation. Heart. 2007;93(6):676–681. doi: 10.1136/hrt.2006.093880.
12. Brescia ST, Rossano JW, Pignatelli R, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. 2013;127(22):2202–2208. doi: 10.1161/circulationaha.113.002511.
13. Xing YL, Ichida F, Matsuoka T, et al.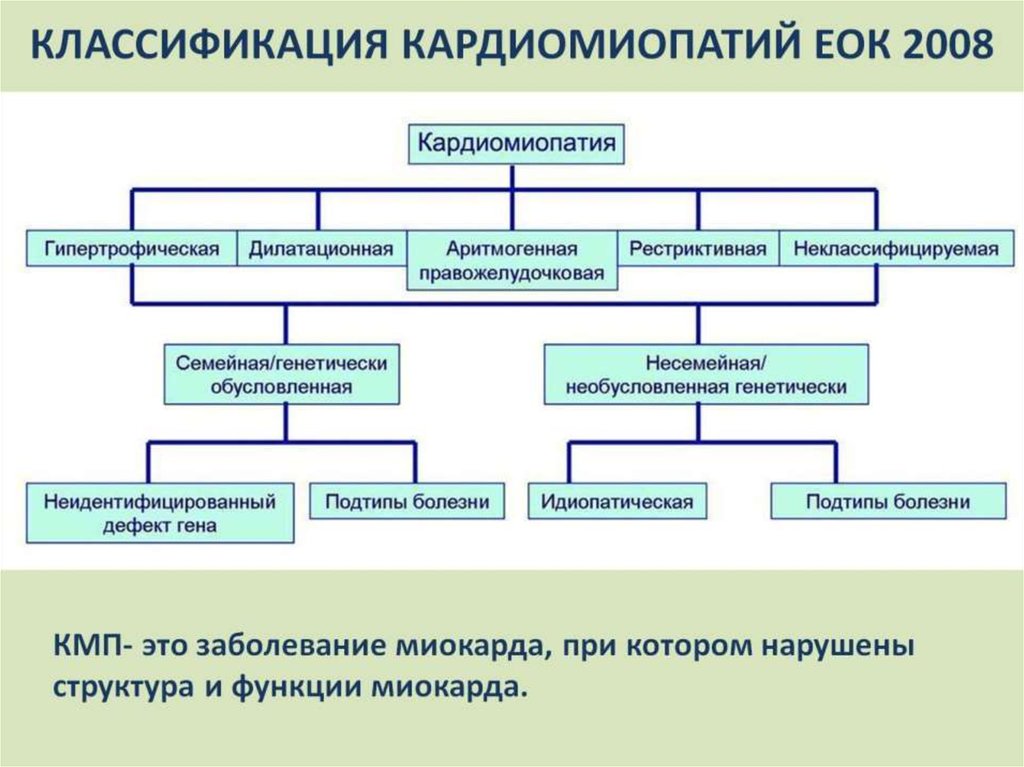 Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88(1):71–77. doi: 10.1016/j.ymgme.2005.11.009.
Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88(1):71–77. doi: 10.1016/j.ymgme.2005.11.009.
14. Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic etiology. Curr Opin Pediatr. 2007;19(6):619–627. doi: 10.1097/MOP.0b013e3282f1ecbc.
15. Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–931. doi: 10.1542/peds.2004-0718.
16. Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J. 2011;32(12):1446–1456. doi: 10.1093/eurheartj/ehq508.
17. Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014;64(17):1840–1850. doi: 10.1016/j.jacc. 2014.08.030.
2014.08.030.
18. Arbustini E, Narula N, Dec GW, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol. 2013;62(22):2046–2072. doi: 10.1016/j.jacc.2013.08.1644.
19. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–1339. doi: 10.1016/j.hrthm.2011.05.020.
20. Хроническая сердечная недостаточность у детей. Клин ические рекомендации. — М.; 2016. Доступно по: http://cardiorus.ru/local/api/download/?id=979b0ebfbd5e6be4f3ceb5d4e414c1b9. Ссылка активна на 10.03.2018.
21. Klaassen S, Probst S, Oechslin E, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117(22):2893–2901. doi: 10.1161/CIRCULATIONAHA.107.746164.
doi: 10.1161/CIRCULATIONAHA.107.746164.
22. Mayosi BM, Khogali S, Zhang B, Watkins H. Cardiac and skeletal actin gene mutations are not a common cause of dilated cardiomyopathy. J Med Genet. 1999;36(10):796–797. doi: 10.1136/jmg.36.10.796.
23. Gajendrarao P, Krishnamoorthy N, Selvaraj S, et al. An investigation of the molecular mechanism of double cMyBP-C mutation in a patient with end-stage hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2015;8(4):232–243. doi: 10.1007/s12265-015-9624-6.
24. Toth T, Nagy V, Faludi R, et al. The Gln1233ter mutation of the myosin binding protein C gene: causative mutation or innocent polymorphism in patients with hypertrophic cardiomyopathy? Int J Cardiol. 2011;153(2):216–219. doi: 10.1016/j.ijcard.2011.09.062.
25. Matsson H, Eason J, Bookwalter CS, et al. Alpha-cardiac actin mutations produce atrial septal defects. Hum Mol Genet. 2008;17(2):256–265. doi: 10.1093/hmg/ddm302.
26. Monserrat L, Hermida-Prieto M, Fernandez X, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28(16):1953–1961. doi: 10.1093/eurheartj/ehm239.
Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28(16):1953–1961. doi: 10.1093/eurheartj/ehm239.
27. Ehlermann P, Weichenhan D, Zehelein J, et al. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med Genet. 2008;9:95. doi: 10.1186/1471-2350-9-95.
28. Hershberger RE, Norton N, Morales A, et al. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3(2):155–161. doi: 10.1161/Circgenetics.109.912345.
29. Probst S, Oechslin E, Schuler P, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4(4): 367–374. doi: 10.1161/CIRCGENETICS.110.959270.
ᐈ Что такое кардиомиопатия? ~【Симптомы, лечение в Киеве】
Кардиомиопатия — это группа сердечных заболеваний, которые вызывают дисфункцию сердца, не позволяя ему снабжать организм кровью. Вначале заболевание может протекать бессимптомно, но по мере прогрессирования возникает недостаточность кровообращения. У 50–60 человек из 100 000 населения развивается кардиопатия. Возникновение болезни может формироваться как первичная патология, когда причина изменений в сердце не полностью известна, и вторичная — на фоне другого заболевания.
Вначале заболевание может протекать бессимптомно, но по мере прогрессирования возникает недостаточность кровообращения. У 50–60 человек из 100 000 населения развивается кардиопатия. Возникновение болезни может формироваться как первичная патология, когда причина изменений в сердце не полностью известна, и вторичная — на фоне другого заболевания.
Классификация кардиомиопатий
Согласно классификации Всемирной организации здравоохранения, выделяются такие виды кардиомиопатий:
- Дилатационная форма — сердце увеличивается в размере в результате чего его стенки истончаются.
- Гипертрофическая форма — стенки сердца становятся толстыми тем самым теряя свою эластичность.
- Рестриктивная форма — самый редкий вид кардиомиопатии. Стенки сердца становятся жесткими, что значительно затрудняет растяжение волокон сердечной мышцы и наполнение его кровью.
Этиология заболевания
Основные причины кардиомиопатии:
- некоторые вирусные, бактериальные, паразитарные инфекции;
- высокое артериальное давление или инфаркт миокарда;
- чрезмерное употребление спиртного;
- некоторые лекарственные препараты, в том числе и некоторые химиотерапевтические препараты.

Другие признаки кардиомиопатии:
- системные заболевания, которые приводят к накоплению некоторых веществ в сердечной мышце и снижают ее эластичность;
- плохо контролируемый сахарный диабет или заболевание щитовидной железы.
Патогенез
В результате развития кардиомиопатии сила сокращения сердца снижается и нарушается работа сердечной мышцы как насоса. Это приводит к недостаточному снабжению организма кислородом. Прогноз зависит от формы заболевания.
Кардиомиопатия: симптомы, причины, клинические проявления
Заболевание чаще всего не проявляется характерными признаками в начальном периоде заболевания.
Только запущенная стадия имеет типичные симптомы кардиомиопатии:
- нерегулярное и учащенное сердцебиение;
- общее беспокойство;
- хроническая усталость;
- отек конечностей;
- головокружение;
- повышенное потоотделение;
- «пятна» перед глазами;
- обморок при физических упражнениях;
- одышка;
- утомительный хронический кашель;
- ощущение нехватки воздуха, невозможность сделать глубокий вдох.

Симптомы кардиомиопатии могут сопровождаться «болью в сердце». Появляется мучительная, давящая, жгучая боль над грудиной, которая отдает в левую руку и лопатку. Длится несколько минут.
Особенности течения заболевания при беременности
Послеродовая кардиомиопатия — редкое, но потенциально смертельное заболевание молодых женщин, которые ранее не получали кардиологическое лечение. Проявляется сердечной недостаточностью, диагностированной в конце беременности или в первые месяцы после родов. Лечение не отличается от стандартной терапии сердечной недостаточности, за исключением ограничений фармакотерапии, характерных для беременности. Необходимо проведение инструментальной диагностики, в основном магнитно-резонансной томографии. Требуется точность в оценке размера полостей сердца для обнаружения тромбов в левом желудочке.
Особенности кардиомиопатий у детей
Что такое кардиомиопатия в детском возрасте? Это прогрессирующая сердечная недостаточность с видимыми симптомами: слабость, снижение толерантности к физической нагрузке, одышка, кашель, боль в груди и обмороки. Наблюдается повышенная склонность к аритмиям сердца и образованию тромбов. При медицинском осмотре обнаруживается увеличение печени, учащенное сердцебиение, учащенное дыхание, похолодание конечностей, гипергидроз, отек и увеличение яремных вен. Детский кардиолог может назначить консервативное лечение. Если оно не дает результата, нужно проводить оперативное лечение.
Наблюдается повышенная склонность к аритмиям сердца и образованию тромбов. При медицинском осмотре обнаруживается увеличение печени, учащенное сердцебиение, учащенное дыхание, похолодание конечностей, гипергидроз, отек и увеличение яремных вен. Детский кардиолог может назначить консервативное лечение. Если оно не дает результата, нужно проводить оперативное лечение.
Осложнения кардиомиопатии
Среди последствий заболевания чаще всего встречаются:
- хроническая сердечная недостаточность;
- аритмии разного характера;
- легочная тромбоэмболия.
Высока вероятность наступления сердечной смерти.
Диагностика заболевания
При установлении диагноза крайне важно отличать кардиомиопатию от других сердечных заболеваний, таких как ишемическая болезнь сердца, гипертония, врожденные аномалии, порок клапана сердца и перикардит. Диагноз ставится на основании клинического обследования, истории болезни и дополнительных анализов.
Подтвердить диагноз помогает эхокардиографическое исследование, которое предоставляет информацию о структуре и функции сердечной мышцы.
Диагностика кардиомиопатии дополнительно базируется на таких исследованиях:
- рентгене грудной клетки;
- анализах состава крови;
- определении уровня гормонов;
- холтере ЭКГ;
- биопсии сердечной мышцы.
Лечение кардиомиопатий
В начальный период диагностики и лечения кардиомиопатии необходимо пребывание в стационаре. Дальнейшая терапия, в зависимости от тяжести заболевания, может проводиться в амбулаторных условиях.
Физическая активность зависит от типа и степени тяжести: варьируется от почти неограниченной до постельного режима.
Больной должен придерживаться специальной диеты: сократить количество употребляемой поваренной соли, жиров животного происхождения, увеличить количество продуктов, богатых витаминами и минералами.
Медикаментозное лечение кардиомиопатии зависит от формы заболевания.
Основные препараты:
- препараты, используемые при сердечной недостаточности;
- препараты для профилактики образования тромбов.
Иногда имплантируют такие устройства, как кардиостимулятор, или в крайнем случае осуществляют пересадку сердца.
Операции при кардиомиопатии
При запущенных кардиомиопатиях, которые сопровождаются тяжелыми симптомами сердечной недостаточности, трансплантация сердца может быть единственным эффективным методом лечения.
Проблемой хирургического вмешательства есть отсутствие доноров. Поэтому хирургическое лечение кардиомиопатии часто ограничивается уменьшением размера стенок камеры. Операция необходима, когда нужно восстановить или заменить поврежденные клапаны или удалить очень толстую часть сердечной мышцы.
Контроль излеченности
Кардиомиопатия полностью не вылечится, но для уменьшения симптомов необходимо постоянное наблюдение у кардиолога. При некоторых формах болезни можно контролировать процесс и поддерживать хорошее качество жизни. Другие формы трудно поддаются лечению и могут привести к развитию сердечной недостаточности. Тем не менее правильно проведенное лечение и постоянное наблюдение позволяют отложить этот момент во времени.
Другие формы трудно поддаются лечению и могут привести к развитию сердечной недостаточности. Тем не менее правильно проведенное лечение и постоянное наблюдение позволяют отложить этот момент во времени.
Профилактика кардиомиопатии
Чтобы исключить риск появления патологии, требуется соблюдение определенных мер:
- ограничить употребление алкоголя;
- снизить потребление соли;
- вылечить заболевания, которые провоцируют патологию;
- периодически проходить инструментальное обследование сердца;
- выполнять все назначения специалиста;
- уменьшить стресс.
Важно предотвратить ишемическую болезнь сердца. Своевременная установка кардиостимулятора при сердечных заболеваниях поможет предотвратить вероятность наступления внезапной смерти.
Советы и рекомендации
Выживаемость пациентов с кардиомиопатией может быть улучшена за счет раннего выявления заболевания и лечения членов семьи из группы риска, у которых еще нет симптомов. Пациентам, генетически подверженным риску возникновения патологии, рекомендуется соблюдать здоровый образ жизни.
Пациентам, генетически подверженным риску возникновения патологии, рекомендуется соблюдать здоровый образ жизни.
После установления диагноза дилатационной кардиомиопатии пациентам следует избегать физических нагрузок и употребления алкоголя.
Вопрос-Ответ
Сколько живут с кардиомиопатией?
Кардиомиопатия — это поражение сердечной мышцы, которое приводит к сердечной недостаточности, а впоследствии и к аритмии или тахикардии. Пациент с диагнозом «кардиомиопатия» состоит на учете у кардиолога. Согласно статистике, прогноз продолжительности жизни при кардиомиопатии зависит от прогрессирования заболевания и составляет примерно от 2 до 15 лет. Основные причины летального исхода: сердечная недостаточность, тромбофлебит, нарушение ритма сердца.
Что такое кардиомиопатия неуточненная?
Кардиомиопатия — это сердечное заболевание, которое вызывает изменение сердечной мышцы. Болезнь не имеет возраст, пол. Для эффективного лечения необходимо определить форму патологии. Кардиомиопатия неуточненная не позволяет специалисту определить эффективность лечения. Для удобства установления диагноза была принята одна общая классификация кардиомиопатии: гипертрофическая, дилатационная, рестриктивная, специфическая, неклассифицированная.
Кардиомиопатия неуточненная не позволяет специалисту определить эффективность лечения. Для удобства установления диагноза была принята одна общая классификация кардиомиопатии: гипертрофическая, дилатационная, рестриктивная, специфическая, неклассифицированная.
Можно ли заниматься спортом при кардиомиопатии?
Спорт и кардиомиопатия. Кардиологическое обследование дает возможность определить оптимальный уровень нагрузки. Физические упражнения способствуют укреплению сердечной мышцы. Интенсивность занятий регламентируется специальными программами обследований для занятий спортом взрослых и детей.
Статья носит информационно-ознакомительный характер. Пожалуйста, помните: самолечение может вредить вашему здоровью.
Автор статьи:
Алексеенко Елена Ивановна
Зав. терапевтическим отделением на Печерске, врач-кардиолог высшей категории
Эксперт по направлению:
Гершкович Игорь Викторович
Врач-кардиолог, реаниматолог, терапевт высшей категории, кандидат медицинских наук
Какой врач лечит кардиомиопатию?
Чтобы пройти современную диагностику и получить эффективное лечение кардиомиопатии в Киеве, следует обратиться в клинику МЕДИКОМ. В нашем центре опытные кардиологи на основании причин кардиомиопатии установят форму патологии и назначат соответствующее лечение. Индивидуальный подход к каждому пациенту позволяет значительно улучшить прогноз даже при тяжелых вариантах развития болезни. Квалифицированный персонал ждет вас в подразделениях клиники в районах Оболони и Печерска. Обращайтесь!
В нашем центре опытные кардиологи на основании причин кардиомиопатии установят форму патологии и назначат соответствующее лечение. Индивидуальный подход к каждому пациенту позволяет значительно улучшить прогноз даже при тяжелых вариантах развития болезни. Квалифицированный персонал ждет вас в подразделениях клиники в районах Оболони и Печерска. Обращайтесь!
все специалисты
Сертификаты
Отзывы
09.04.2021 18:17
Олександр
Серцебиття, спасибі, завтра біжу до лікаря.
26.02.2021 14:57
Жанна Фатєєва
Пережила хворобу, зараз веду здоровий спосіб життя, тому і відчуваю себе добре.
04.02.2021 19:01
Діна Петрівна
Спасибі сервісу, завжди інформативні статті, багато корисної інформації.
14.01.2021 11:33
Елена Владимировна
С сердцем не шутки, главное вовремя обратится к врачу.
18.11.2020 17:26
Марина Дмитриевна
Хорошая статья. Спасибо.
02.10.2020 13:20
Надежда
Долго болела, наконец выписали, теперь изменю свои привычки. Отличная статья.
Отличная статья.
Показать еще 3
Всего 6 отзывов
оставить отзыв
Общие сведения о кардиомиопатиях у детей
Введение
Детские кардиомиопатии представляют собой необычную и гетерогенную группу заболеваний, характеризующихся структурными, механическими и электрическими нарушениями сердечной мышцы (1, 2). Этиология разнообразна и включает инфекции, воздействие токсинов, тахиаритмии, генетические мутации и лежащие в их основе метаболические или нервно-мышечные нарушения (1–6). Большие популяционные регистры вместе с национальными и многоцентровыми исследованиями внесли значительный вклад в увеличение знаний об эпидемиологии и исходах детских кардиомиопатий (5, 7–24). Общая ежегодная заболеваемость оценивается примерно в 1 случай на 100 000 детей со значительно более высокой заболеваемостью в течение первых 2 лет жизни (7, 17, 18). Дилатационная и гипертрофическая кардиомиопатии являются наиболее распространенными типами, тогда как неуплотненная и рестриктивная кардиомиопатия левого желудочка встречаются реже (7, 17). Аритмогенная желудочковая кардиомиопатия редко диагностируется в детском возрасте и не будет обсуждаться в этой статье. Термины «расширенный», «гипертрофический», «рестриктивный» и «некомпактный» обозначают различные фенотипы (см. рис. 1) и, таким образом, помогают группировать кардиомиопатии, однако они не описывают конкретные формы заболевания. Европейское общество кардиологов классифицирует кардиомиопатии в соответствии с их преобладающим фенотипом, но не признает некомпактность левого желудочка как отдельную нозологию, в то время как Американская ассоциация кардиологов классифицирует кардиомиопатии в соответствии с их этиологией, при этом некомпактность левого желудочка рассматривается как отдельная болезнь. сущность (25, 26). У некоторых детей может быть смешанный фенотип, а фенотипы кардиомиопатии могут быть волнообразными или трансформироваться в течение болезни (1, 12, 13, 27). В этом обзоре обобщены наиболее распространенные формы детских кардиомиопатий с акцентом на эпидемиологию и естественное течение.
Аритмогенная желудочковая кардиомиопатия редко диагностируется в детском возрасте и не будет обсуждаться в этой статье. Термины «расширенный», «гипертрофический», «рестриктивный» и «некомпактный» обозначают различные фенотипы (см. рис. 1) и, таким образом, помогают группировать кардиомиопатии, однако они не описывают конкретные формы заболевания. Европейское общество кардиологов классифицирует кардиомиопатии в соответствии с их преобладающим фенотипом, но не признает некомпактность левого желудочка как отдельную нозологию, в то время как Американская ассоциация кардиологов классифицирует кардиомиопатии в соответствии с их этиологией, при этом некомпактность левого желудочка рассматривается как отдельная болезнь. сущность (25, 26). У некоторых детей может быть смешанный фенотип, а фенотипы кардиомиопатии могут быть волнообразными или трансформироваться в течение болезни (1, 12, 13, 27). В этом обзоре обобщены наиболее распространенные формы детских кардиомиопатий с акцентом на эпидемиологию и естественное течение.
Рисунок 1 . Эхокардиографические изображения фенотипов кардиомиопатии. Верхушечные четырехкамерные проекции, демонстрирующие (A) расширенные левый желудочек и левое предсердие у пациента с ДКМП, (B) гипертрофию межжелудочковой перегородки и свободной стенки левого желудочка у пациента с ГКМП, (C) массивно расширенные предсердия и небольшие полости правого и левого желудочка у пациента с ОКМ, (D) обширно трабекулярный миокард с уплотненным и неуплотненным слоем и глубокими межтрабекулярными углублениями, наиболее заметными в области верхушки левого желудочка и свободной стенки у пациента с НМЛЖ.
Дилатационная кардиомиопатия
Этиология
Дилатационная кардиомиопатия (ДКМП) характеризуется дилатацией левого желудочка и систолической дисфункцией. Важные этиологии в детском возрасте включают инфекции, токсические причины (включая химиотерапию), генетические мутации и другие причины, такие как врожденные нарушения метаболизма и нервно-мышечные расстройства (1). Недавние достижения в генетической диагностике, в том числе внедрение технологий секвенирования ДНК следующего поколения и расширенных панелей генов кардиомиопатии, увеличили частоту обнаружения патогенных мутаций у взрослых пациентов с ДКМП примерно до 40% (28, 29).). Считается, что мутации гена саркомера ответственны за 35–40% генетических случаев (1). Признаки миокардита были обнаружены примерно у трети детей с ДКМП, подвергшихся ранней эндомиокардиальной биопсии (20, 21, 30). Люди с вариантами генов, кодирующих структурные белки сердца, могут быть особенно восприимчивы к тяжелому миокардиту (31, 32). Однако, несмотря на значительный прогресс в диагностике за последнее десятилетие, этиология ДКМП у детей часто остается неизвестной.
Недавние достижения в генетической диагностике, в том числе внедрение технологий секвенирования ДНК следующего поколения и расширенных панелей генов кардиомиопатии, увеличили частоту обнаружения патогенных мутаций у взрослых пациентов с ДКМП примерно до 40% (28, 29).). Считается, что мутации гена саркомера ответственны за 35–40% генетических случаев (1). Признаки миокардита были обнаружены примерно у трети детей с ДКМП, подвергшихся ранней эндомиокардиальной биопсии (20, 21, 30). Люди с вариантами генов, кодирующих структурные белки сердца, могут быть особенно восприимчивы к тяжелому миокардиту (31, 32). Однако, несмотря на значительный прогресс в диагностике за последнее десятилетие, этиология ДКМП у детей часто остается неизвестной.
Эпидемиология
ДКМП является наиболее распространенным типом детской кардиомиопатии. Он включал около половины всех случаев в Реестре детской кардиомиопатии (PCMR), крупном многоцентровом североамериканском исследовании, а также в Национальном австралийском исследовании детской кардиомиопатии (NACCS), популяционном когортном исследовании, в которое были включены дети младше 10 лет. возраста на момент постановки диагноза. Общая заболеваемость составила 0,58–0,73 на 100 000 детей и значительно варьировала в зависимости от возраста (см. табл. 1). В обоих исследованиях самая высокая годовая заболеваемость наблюдалась в течение первого года жизни (см. Таблицу 1) (7, 17). Общенациональное исследование кардиомиопатии в Финляндии, которое включало только случаи идиопатической кардиомиопатии, продемонстрировало общую частоту ДКМП 0,34 на 100 000 детей в возрасте до 20 лет и 11-кратное увеличение заболеваемости в течение первого года жизни (см. Таблицу 1) (18). В большом проспективном исследовании, проведенном в Соединенном Королевстве и Ирландии, оценивалась заболеваемость впервые возникшей сердечной недостаточностью у детей из-за заболеваний сердечной мышцы известной и неизвестной этиологии. Авторы сообщили о частоте 0,76 симптоматических случаев ДКМП на 100 000 детей в возрасте до 16 лет (19).). Лимфоцитарный миокардит составил 22% всех случаев в этом исследовании и составил 16–36% случаев ДКМП в PCMR и NACCS (19–21).
возраста на момент постановки диагноза. Общая заболеваемость составила 0,58–0,73 на 100 000 детей и значительно варьировала в зависимости от возраста (см. табл. 1). В обоих исследованиях самая высокая годовая заболеваемость наблюдалась в течение первого года жизни (см. Таблицу 1) (7, 17). Общенациональное исследование кардиомиопатии в Финляндии, которое включало только случаи идиопатической кардиомиопатии, продемонстрировало общую частоту ДКМП 0,34 на 100 000 детей в возрасте до 20 лет и 11-кратное увеличение заболеваемости в течение первого года жизни (см. Таблицу 1) (18). В большом проспективном исследовании, проведенном в Соединенном Королевстве и Ирландии, оценивалась заболеваемость впервые возникшей сердечной недостаточностью у детей из-за заболеваний сердечной мышцы известной и неизвестной этиологии. Авторы сообщили о частоте 0,76 симптоматических случаев ДКМП на 100 000 детей в возрасте до 16 лет (19).). Лимфоцитарный миокардит составил 22% всех случаев в этом исследовании и составил 16–36% случаев ДКМП в PCMR и NACCS (19–21). Эндомиокардиальная биопсия не всегда выполняется в впервые диагностированных случаях ДКМП, и наличие положительных результатов гистологического исследования лимфоцитарного миокардита быстро уменьшается в течение нескольких недель после поступления, что может привести к недооценке воспалительного ДКМП у детей (21, 33).
Эндомиокардиальная биопсия не всегда выполняется в впервые диагностированных случаях ДКМП, и наличие положительных результатов гистологического исследования лимфоцитарного миокардита быстро уменьшается в течение нескольких недель после поступления, что может привести к недооценке воспалительного ДКМП у детей (21, 33).
Таблица 1 . Годовая заболеваемость и средний возраст при постановке диагноза для каждого типа кардиомиопатии (КМ).
Особенности клинического течения
Большинство детей с ДКМП, особенно младенцы, имеют застойную сердечную недостаточность различной степени тяжести с симптомами, варьирующими от затруднений при кормлении до сердечно-сосудистого коллапса и редко внезапной смерти. Детям более старшего возраста с положительным семейным анамнезом, а также детям с нервно-мышечными расстройствами или врожденными нарушениями обмена веществ может быть поставлен диагноз в рамках рутинного скрининга до появления симптомов. Лимфоцитарный миокардит имеет фенотип, который может быть неотличим от идиопатического ДКМП. История предшествующего вирусного заболевания не всегда присутствует и может вводить в заблуждение (34). Данные PCMR и NACCS показали, что на момент постановки диагноза 71–90% детей с ДКМП имели клинические признаки застойной сердечной недостаточности (20, 21). В когорте NACCS DCM 5% имели внезапную сердечную смерть, 2% — непереносимость физической нагрузки или аритмию и 3% — рутинный семейный скрининг. Госпитализация в отделение интенсивной терапии потребовалась в 45% случаев, а инотропная поддержка проводилась в 40% случаев (21). Серьезные сопутствующие заболевания, связанные с госпитализацией в отделение интенсивной терапии, связанной с кардиомиопатией, включали почечную недостаточность, тромбоэмболические осложнения и печеночную недостаточность (35).
История предшествующего вирусного заболевания не всегда присутствует и может вводить в заблуждение (34). Данные PCMR и NACCS показали, что на момент постановки диагноза 71–90% детей с ДКМП имели клинические признаки застойной сердечной недостаточности (20, 21). В когорте NACCS DCM 5% имели внезапную сердечную смерть, 2% — непереносимость физической нагрузки или аритмию и 3% — рутинный семейный скрининг. Госпитализация в отделение интенсивной терапии потребовалась в 45% случаев, а инотропная поддержка проводилась в 40% случаев (21). Серьезные сопутствующие заболевания, связанные с госпитализацией в отделение интенсивной терапии, связанной с кардиомиопатией, включали почечную недостаточность, тромбоэмболические осложнения и печеночную недостаточность (35).
Естественная история
Исходы для детей с ДКМП сильно различаются: от полного выздоровления до смерти или необходимости трансплантации. За пределами младенческого возраста ДКМП является наиболее частым показанием к трансплантации сердца у детей (36). Данные PCMR и NACCS продемонстрировали общую 5-летнюю выживаемость без трансплантации 54–65 % (см. Таблицу 2) (20, 21). Стратификация риска помогает выявить лиц, которым можно оказать медикаментозное лечение, и тех, кому требуется расширенная терапия сердечной недостаточности, включая внесение в список лиц, нуждающихся в трансплантации сердца. Анализ выживаемости без трансплантации в соответствии с основной этиологией из PCMR показан в таблице 2 (20). Данные о долгосрочных исходах из NACCS показали, что выживаемость без трансплантации составляет 56% через 20 лет (22). Самый высокий риск наблюдался сразу после постановки диагноза: 26% риска смерти или трансплантации в течение первого года и только 1% в год после этого (22). Сходные результаты были получены в британском исследовании детей, поступивших с впервые возникшей сердечной недостаточностью на фоне дилатационной кардиомиопатии. Годичная бессобытийная выживаемость составила всего 66, а 16% перенесли трансплантацию сердца в течение первого года после постановки диагноза (19).
Данные PCMR и NACCS продемонстрировали общую 5-летнюю выживаемость без трансплантации 54–65 % (см. Таблицу 2) (20, 21). Стратификация риска помогает выявить лиц, которым можно оказать медикаментозное лечение, и тех, кому требуется расширенная терапия сердечной недостаточности, включая внесение в список лиц, нуждающихся в трансплантации сердца. Анализ выживаемости без трансплантации в соответствии с основной этиологией из PCMR показан в таблице 2 (20). Данные о долгосрочных исходах из NACCS показали, что выживаемость без трансплантации составляет 56% через 20 лет (22). Самый высокий риск наблюдался сразу после постановки диагноза: 26% риска смерти или трансплантации в течение первого года и только 1% в год после этого (22). Сходные результаты были получены в британском исследовании детей, поступивших с впервые возникшей сердечной недостаточностью на фоне дилатационной кардиомиопатии. Годичная бессобытийная выживаемость составила всего 66, а 16% перенесли трансплантацию сердца в течение первого года после постановки диагноза (19). ).
).
Таблица 2 . Выживаемость без трансплантации для каждого типа кардиомиопатии (КМ).
Другие факторы риска у детей с ДКМП связаны с возрастом на момент постановки диагноза и тяжестью сердечной дисфункции. Факторы риска смерти или трансплантации в NACCS включали возраст младше 4 недель или старше 5 лет на момент обращения, семейную дилатационную кардиомиопатию, более низкое исходное фракционное укорочение z -баллов и отсутствие улучшения фракционного укорочения z -баллов во время наблюдения (21, 22). Благоприятные исходы, в том числе безтрансплантационная выживаемость и обратное ремоделирование, чаще встречались у детей с лимфоцитарным миокардитом по сравнению с детьми другой этиологии, при этом эхокардиографическая нормализация функции ЛЖ была обнаружена в 9 случаях.2% во время последующего наблюдения (22). Хотя реже, обратное моделирование также наблюдалось у детей без подтвержденного или подозреваемого миокардита. Эверитт и др. сообщили об эхокардиографическом восстановлении функции ЛЖ в течение 2 лет в 22% случаев с идиопатической ДКМП, включенной в ПКМР. Младший возраст (<10 лет) и меньшая дилатация ЛЖ на момент постановки диагноза были независимыми прогностическими факторами нормализации эхокардиографии. У некоторых пациентов развился рецидив застойной сердечной недостаточности после первоначальной нормализации эхокардиографии (37). Было обнаружено, что сохранение повышенных уровней N-концевого про-МНП после начальной стабилизации предсказывает риск сердечной смерти у детей с идиопатической ДКМП (38).
Младший возраст (<10 лет) и меньшая дилатация ЛЖ на момент постановки диагноза были независимыми прогностическими факторами нормализации эхокардиографии. У некоторых пациентов развился рецидив застойной сердечной недостаточности после первоначальной нормализации эхокардиографии (37). Было обнаружено, что сохранение повышенных уровней N-концевого про-МНП после начальной стабилизации предсказывает риск сердечной смерти у детей с идиопатической ДКМП (38).
Пал и др. рассмотрели факторы риска внезапной сердечной смерти (ВСС) при ДКМП из PCMR и обнаружили, что 5-летняя заболеваемость составляет 3%. Младший возраст при постановке диагноза (<14,3 лет), систолическая дилатация ЛЖ (конечный систолический размер ЛЖ х — балл >2,6) и истончение задней стенки были идентифицированы как факторы риска внезапной смерти (39).
Общая выживаемость при ДКМП у детей в Северной Америке улучшилась в последнее время, несмотря на то, что показатели эхокардиографической нормализации и трансплантации сердца не отличались по сравнению с предыдущим периодом (40).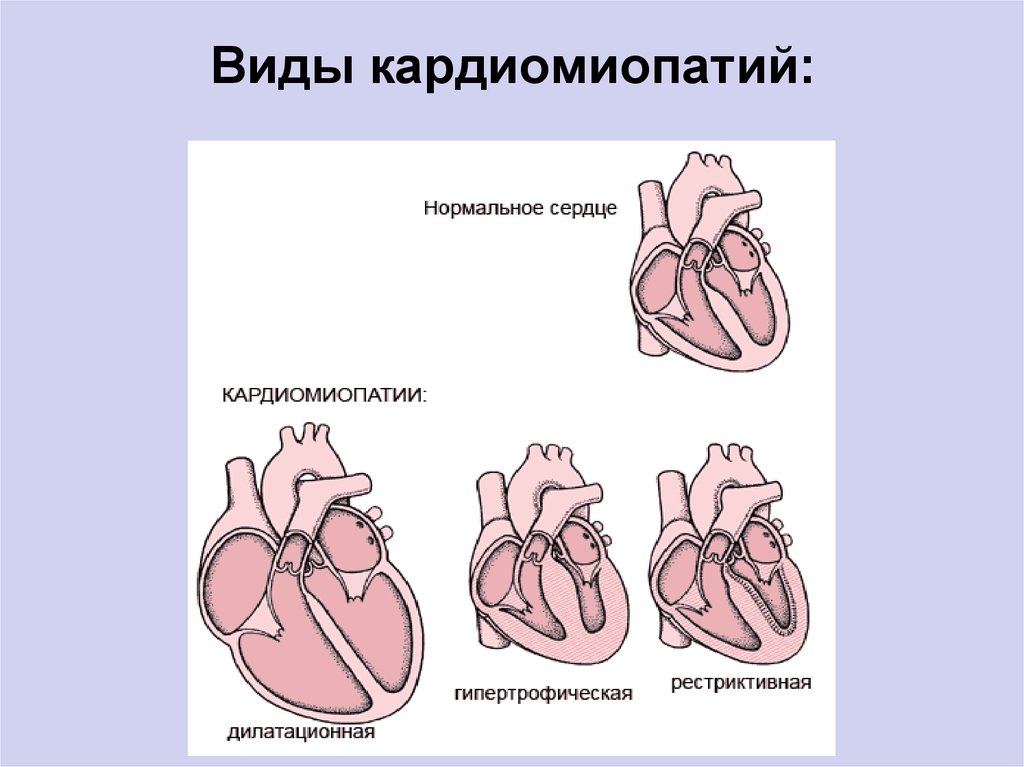 Это может быть связано с улучшением реанимации и/или использованием терапии хронической сердечной недостаточности у взрослых.
Это может быть связано с улучшением реанимации и/или использованием терапии хронической сердечной недостаточности у взрослых.
Гипертрофическая кардиомиопатия
Этиология
Гипертрофическая кардиомиопатия (ГКМП) представляет собой состояние, характеризующееся гипертрофией левого или бивентрикулярного отдела сердца при отсутствии структурного заболевания сердца или повышенной постнагрузки на желудочки. Саркомерные мутации представляют собой наиболее распространенную генетическую причину у детей и взрослых с частотой обнаружения около 60% при детской ГКМП, диагностированной после первого года жизни, аналогично таковой у взрослых пациентов с ГКМП (41). По сравнению с ГКМП у взрослых, детская ГКМП представляет собой гораздо более гетерогенную группу с различной этиологией и спектром заболеваний (6). К генетическим причинам относятся врожденные нарушения метаболизма, синдромы пороков развития (в первую очередь РАсопатии), нервно-мышечные заболевания, а также патогенные мутации в генах, кодирующих саркомерные белки. Типичными причинами внутри этих четырех категорий являются болезнь Помпе, синдром Нунана, атаксия Фридрейха и мутации MYBPC3 или MYH7 соответственно (1, 23).
Типичными причинами внутри этих четырех категорий являются болезнь Помпе, синдром Нунана, атаксия Фридрейха и мутации MYBPC3 или MYH7 соответственно (1, 23).
Эпидемиология
ГКМП составляет вторую по частоте группу детских кардиомиопатий, составляющую 25–42% всех случаев соответственно. Общая годовая заболеваемость составляет 0,24–0,47 на 100 000 детей (см. табл. 1) (7, 17, 18). ГКМП, вызванная врожденными нарушениями обмена веществ и синдромами пороков развития, обычно проявляется в младенчестве и вносит значительный вклад в ранний первый пик заболеваемости. Второй, меньший пик в подростковом и раннем взрослом возрасте в значительной степени связан с мутациями саркомерных белков (1, 41). Данные NACCS, в которые были включены дети только в возрасте 0–10 лет на момент постановки диагноза и исключены дети с метаболическими и нервно-мышечными заболеваниями, показали, что средний возраст при постановке диагноза составляет 5,7 месяца. NACCS и PCMR сообщают о заметном снижении заболеваемости между первым и последующими годами жизни (см. Таблицу 1). В финском исследовании, в которое также были исключены пациенты с метаболической и нервно-мышечной этиологией, но были включены дети в возрасте до 20 лет на момент постановки диагноза, средний возраст на момент постановки диагноза составил 13 лет, а 39 лет% пациентов были старше 15 лет на момент поступления (см. Таблицу 1).
Таблицу 1). В финском исследовании, в которое также были исключены пациенты с метаболической и нервно-мышечной этиологией, но были включены дети в возрасте до 20 лет на момент постановки диагноза, средний возраст на момент постановки диагноза составил 13 лет, а 39 лет% пациентов были старше 15 лет на момент поступления (см. Таблицу 1).
Особенности клинического течения
Клиническое состояние при появлении варьируется от бессимптомного до симптомов непереносимости физической нагрузки, боли в груди, сердцебиения, обморока или остановки сердца (1). Застойная сердечная недостаточность или аритмические симптомы обнаруживаются в 10–15% случаев при поступлении (6, 24). Дети с врожденными нарушениями обмена веществ и синдромами пороков развития обычно выявляются раньше и с большей вероятностью имеют застойную сердечную недостаточность на момент постановки диагноза (23). Прерванная внезапная сердечная смерть или внебольничная остановка являются необычными начальными проявлениями ГКМП в детском возрасте (6).
Естественная история
Естественная история и исход детской ГКМП в значительной степени зависят от возраста на момент поступления и лежащей в основе этиологии. Самый высокий риск смертности наблюдается у лиц, диагностированных в течение первого года жизни (24).
Общая выживаемость без смерти или трансплантации составила около 90% через 5 лет и 78% через 20 лет после поступления (см. Таблицу 2) (6, 8, 24). Риск смерти или трансплантации составлял 14% в течение первого года после поступления, снижаясь до 0,4% в последующие годы (8).
Наихудшие исходы наблюдаются у младенцев с сердечной недостаточностью на момент постановки диагноза и у детей более старшего возраста с выраженной рестриктивной патофизиологией. Другие факторы риска включают концентрическую гипертрофию левого желудочка при постановке диагноза, синдром Нунан и увеличение толщины свободной стенки левого желудочка и ухудшение систолической функции левого желудочка во время наблюдения (8). В PCMR дети с несиндромальной ГКМП, диагностированной до 1 года, имели более высокую смертность, которая снижалась у выживших в младенчестве (23).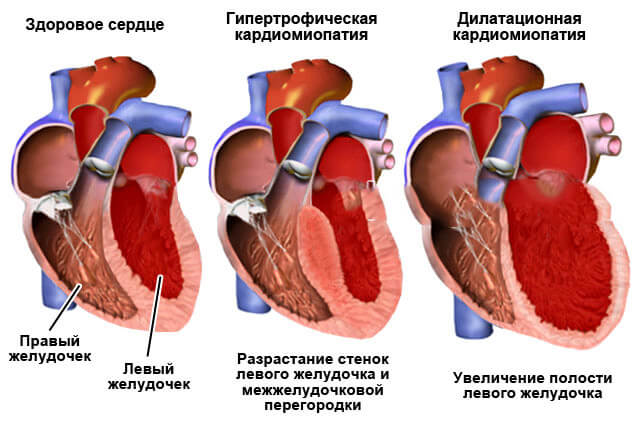 Точно так же у пациентов с синдромом Нунана наблюдалось заметное снижение годовой выживаемости при диагностированной застойной сердечной недостаточности в возрасте до 6 месяцев (42). Самая высокая 5-летняя выживаемость наблюдалась при ГКМП, вторичной по отношению к нервно-мышечному заболеванию (см. Таблицу 2) (23).
Точно так же у пациентов с синдромом Нунана наблюдалось заметное снижение годовой выживаемости при диагностированной застойной сердечной недостаточности в возрасте до 6 месяцев (42). Самая высокая 5-летняя выживаемость наблюдалась при ГКМП, вторичной по отношению к нервно-мышечному заболеванию (см. Таблицу 2) (23).
В то время как застойная сердечная недостаточность является причиной большинства ранних смертей при ГКМП у детей, наиболее распространенным видом смерти в целом является ВСС (6, 8). Аритмические события наблюдались с частотой 1,2 на 100 пациенто-лет в большом исследовании в Великобритании, с более частым возникновением у несиндромальных пациентов (6). Выявление специфических педиатрических факторов риска ВСС имеет важное значение для определения имплантируемого кардиовертера-дефибриллятора (ИКД) у населения, которое часто не испытывает сердечных симптомов в повседневной жизни. Систематический обзор и метаанализ клинических факторов риска внезапной сердечной смерти при детской кардиомиопатии выявили предшествующие неблагоприятные сердечные события, неустойчивую желудочковую тахикардию, обмороки и крайнюю гипертрофию левого желудочка в качестве основных факторов (43). Норриш и др. недавно описали новую модель прогнозирования риска ВСС при ГКМП у детей с целью предоставления индивидуальных оценок риска. Обнаружено, что необъяснимый обморок, максимальная толщина стенки левого желудочка, диаметр левого предсердия и неустойчивая ЖТ имеют самую сильную связь с комбинированным исходом ВСС или эквивалентным событием (44). Мирон и др. также описали модель прогнозирования риска ВСС для педиатрической ГКМП. Эта группа использовала вышеупомянутые четыре фактора риска, а также возраст на момент постановки диагноза для клинической модели и добавила наличие варианта патогенного гена для комбинированной клинической/генетической модели (45). Взаимосвязь между обструкцией оттока левого желудочка и внезапной смертью сложна, при этом некоторые исследования показывают либо защитный эффект, либо обратную зависимость у детей с самыми высокими градиентами (44–46). Аритмические события, приводящие к внезапной сердечной смерти, продолжают происходить во взрослых когортах с частотой около 0,7% в год (47).
Норриш и др. недавно описали новую модель прогнозирования риска ВСС при ГКМП у детей с целью предоставления индивидуальных оценок риска. Обнаружено, что необъяснимый обморок, максимальная толщина стенки левого желудочка, диаметр левого предсердия и неустойчивая ЖТ имеют самую сильную связь с комбинированным исходом ВСС или эквивалентным событием (44). Мирон и др. также описали модель прогнозирования риска ВСС для педиатрической ГКМП. Эта группа использовала вышеупомянутые четыре фактора риска, а также возраст на момент постановки диагноза для клинической модели и добавила наличие варианта патогенного гена для комбинированной клинической/генетической модели (45). Взаимосвязь между обструкцией оттока левого желудочка и внезапной смертью сложна, при этом некоторые исследования показывают либо защитный эффект, либо обратную зависимость у детей с самыми высокими градиентами (44–46). Аритмические события, приводящие к внезапной сердечной смерти, продолжают происходить во взрослых когортах с частотой около 0,7% в год (47).
Рестриктивная кардиомиопатия
Этиология
Рестриктивная кардиомиопатия (РКМ) представляет собой редкую форму заболевания сердечной мышцы, определяемую как «нормальный или уменьшенный объем обоих желудочков, связанный с биатриальным расширением, нормальной толщиной стенки левого желудочка и атриовентрикулярных клапанов, нарушением наполнения желудочков с ограничительная физиология и нормальная (или почти нормальная) систолическая функция» (26). Почти у четверти пациентов с РКМ имеется семейный анамнез кардиомиопатии (10). Сообщалось об увеличении числа генетических мутаций в саркомерных и несаркомерных белках, что свидетельствует о совпадении с другими формами кардиомиопатии (5, 48, 49).). Значительная подгруппа случаев РКМП имеет смешанный фенотип, чаще всего сочетающий характеристики РКМП и ГКМП (10). РКМ также был описан в связи с врожденными нарушениями метаболизма, инфильтративным заболеванием и скелетной миопатией (1, 50).
Эпидемиология
РКМ — самая редкая форма детской кардиомиопатии с частотой 0,03–0,04 на 100 000 детей в Австралии и США (см.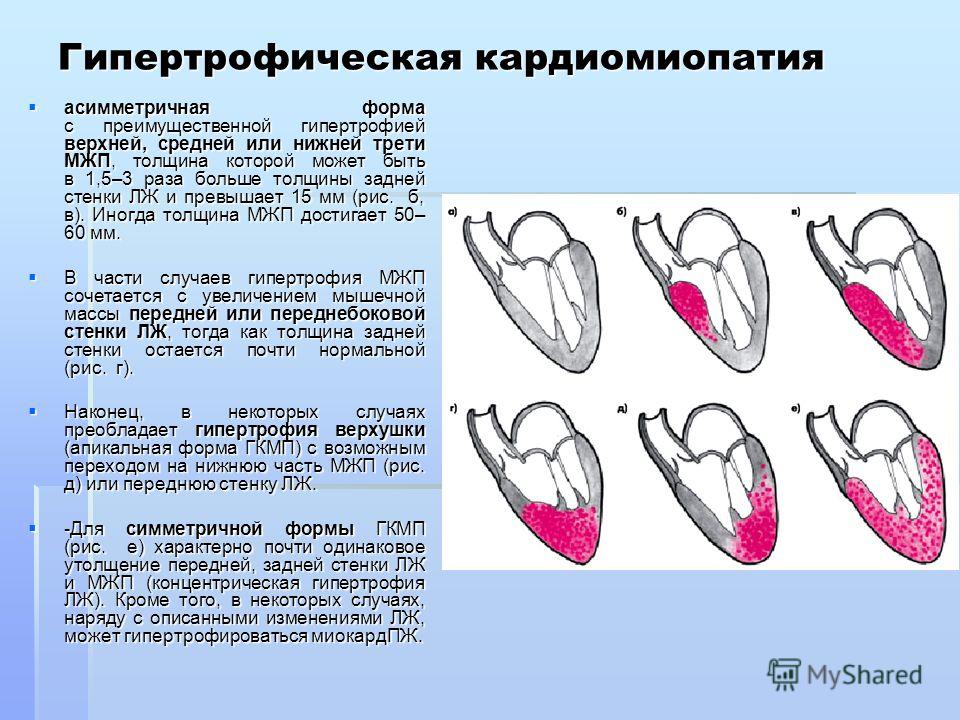 табл. 1) (7, 17). На долю RCM приходилось 2,5% случаев в NACCS, а в PCMR сообщалось о 3% случаев чистого RCM и дополнительно о 1,5% случаев смешанного фенотипа RCM/HCM (7, 17). Возраст при постановке диагноза колеблется от раннего младенчества до позднего взросления (1). В отличие от других детских кардиомиопатий, РКМП становится более частым с возрастом. Только 10% случаев чистой РКМ в ПКМР были диагностированы в течение первого года жизни (10).
табл. 1) (7, 17). На долю RCM приходилось 2,5% случаев в NACCS, а в PCMR сообщалось о 3% случаев чистого RCM и дополнительно о 1,5% случаев смешанного фенотипа RCM/HCM (7, 17). Возраст при постановке диагноза колеблется от раннего младенчества до позднего взросления (1). В отличие от других детских кардиомиопатий, РКМП становится более частым с возрастом. Только 10% случаев чистой РКМ в ПКМР были диагностированы в течение первого года жизни (10).
Проявления
Ранние симптомы РКМ могут быть неспецифическими, включая общую усталость и непереносимость физической нагрузки. Клинические признаки, вторичные по отношению к повышенному системному и легочному венозному давлению, включают периферические отеки, гепатомегалию, отек легких и легочную гипертензию (1). На более поздних стадиях заболевания у пациентов может развиться систолическая дисфункция (1). Обморок — неспецифический, но зловещий симптом, который может быть вызван аритмией, коронарной ишемией или тромбоэмболическими явлениями (51).
Естественная история
В то время как ОКМ имеет наихудшие исходы среди всех детских кардиомиопатий, естественное течение заболевания в значительной степени затемняется ранним направлением на трансплантацию. Поэтому данных о долгосрочной выживаемости без трансплантации немного. Из-за его неумолимого и прогрессирующего характера у пациентов существует риск внезапной смерти, застойной сердечной недостаточности, предсердных и желудочковых аритмий, нарушений проводимости и тромбоэмболии (1, 52). Выживаемость без трансплантации для детей с чистой РКМ в ПЦМР составила 48 и 22% через 1 и 5 лет после установления диагноза соответственно (10). В группе со смешанным фенотипом (ОКМ/ГКМ) 5-летняя выживаемость без трансплантации была в 2 раза выше (см. Таблицу 2). Общая свобода от смерти через 5 лет была одинаковой для обеих когорт, что указывает на предпочтение более ранней трансплантации у пациентов с чистой РКМ (10). Руссо и др. рассмотрели 21 случай РКМ в одном центре ретроспективного анализа и сообщили о выживаемости без трансплантации 80,5 и 20% через 1 и 10 лет соответственно (53).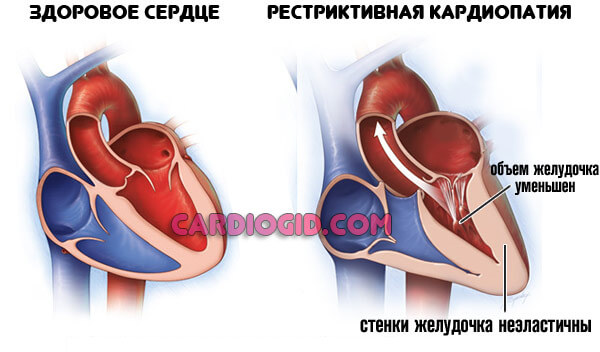 Андерсон и др. проанализировали их опыт работы в учреждениях для детей с ОКМ, сравнивая историческую когорту из 9случаев, диагностированных между 1975 и 1993 годами, с современной когортой из 12 случаев. Выживаемость без трансплантации в течение 5 лет составила 38% в обеих группах, однако общая выживаемость в современной группе составила 80 против 38% в исторической группе (54).
Андерсон и др. проанализировали их опыт работы в учреждениях для детей с ОКМ, сравнивая историческую когорту из 9случаев, диагностированных между 1975 и 1993 годами, с современной когортой из 12 случаев. Выживаемость без трансплантации в течение 5 лет составила 38% в обеих группах, однако общая выживаемость в современной группе составила 80 против 38% в исторической группе (54).
Из-за нечастости ОКМ у детей и небольших групп исследования оценка факторов риска для исхода оказалась сложной, а результаты были противоречивыми. В PCMR симптомы сердечной недостаточности и более низкое фракционное укорочение z баллов при постановке диагноза были идентифицированы как независимые факторы риска снижения выживаемости без трансплантации (10). Точно так же более высокие исходные эхокардиографические размеры левого предсердия и потребность в диуретиках во время последующего наблюдения были связаны с повышенной смертностью (49).). Андерсон и др. наблюдали связь заметного повышения допплеровского соотношения E/e’ митрального клапана на эхокардиографии с повышенной смертностью (54). Ривенс и др. оценили факторы риска, предсказывающие внезапную смерть и сердечно-сосудистый коллапс, у 18 детей с ОКМ из одноцентрового ретроспективного исследования. Они сообщили о повышенном риске связанных с ишемией осложнений и смертности во всей группе пациентов. Риск внезапной смерти был самым высоким у девочек с клиническими признаками, указывающими на ишемию, в частности с болью в груди и обмороком при поступлении. Подгруппа с риском внезапной смерти выглядела хорошо и не имела клинических признаков продолжающейся застойной сердечной недостаточности (51). Уолш и др. наблюдали, что удлинение PR и более широкий комплекс QRS на исходной ЭКГ были связаны с увеличением частоты острых сердечных событий, и они обнаружили значительный риск острой блокады сердца высокой степени у пациентов с ОКМ (52). Повышенное легочное сосудистое сопротивление присутствует у 40% детей с РКМ и может влиять на сроки направления на трансплантацию (11). Серийная катетеризация сердца часто проводится для выявления этого серьезного осложнения, которое может повлиять на пригодность трансплантата (5, 11).
Ривенс и др. оценили факторы риска, предсказывающие внезапную смерть и сердечно-сосудистый коллапс, у 18 детей с ОКМ из одноцентрового ретроспективного исследования. Они сообщили о повышенном риске связанных с ишемией осложнений и смертности во всей группе пациентов. Риск внезапной смерти был самым высоким у девочек с клиническими признаками, указывающими на ишемию, в частности с болью в груди и обмороком при поступлении. Подгруппа с риском внезапной смерти выглядела хорошо и не имела клинических признаков продолжающейся застойной сердечной недостаточности (51). Уолш и др. наблюдали, что удлинение PR и более широкий комплекс QRS на исходной ЭКГ были связаны с увеличением частоты острых сердечных событий, и они обнаружили значительный риск острой блокады сердца высокой степени у пациентов с ОКМ (52). Повышенное легочное сосудистое сопротивление присутствует у 40% детей с РКМ и может влиять на сроки направления на трансплантацию (11). Серийная катетеризация сердца часто проводится для выявления этого серьезного осложнения, которое может повлиять на пригодность трансплантата (5, 11).
Некомпактность левого желудочка
Этиология
Некомпактность левого желудочка (LVNC) представляет собой гетерогенную форму кардиомиопатии, характеризующуюся чрезмерной трабекуляцией левого или обоих желудочков с глубокими межтрабекулярными углублениями, чаще всего поражающей верхушку левого желудочка. LVNC был классифицирован Американской ассоциацией сердца в 2006 г. как отдельная кардиомиопатия (26), однако продолжается дискуссия о том, является ли он отдельной единицей или морфологическим фенотипом (55). Остановка нормального эндомиокардиального морфогенеза с отсутствием уплотнения трабекул считается причиной, особенно в педиатрических случаях (3). Подобный фенотип может проявиться в любое время во взрослой жизни вследствие состояний, связанных с повышенной преднагрузкой левого желудочка (56). LVNC может быть изолирован или связан с другими фенотипами кардиомиопатии, аритмиями или врожденными пороками сердца. LVNC обычно встречается при синдроме Барта, Х-сцепленном рецессивном заболевании, вызванном мутациями гена тафаззина, а также у пациентов с врожденными нарушениями метаболизма, нервно-мышечными заболеваниями и генетическими синдромами (1). Генетическое тестирование выявляет варианты в 30–45% случаев, причем чаще всего обнаруживаются саркомерные мутации (3, 56).
Генетическое тестирование выявляет варианты в 30–45% случаев, причем чаще всего обнаруживаются саркомерные мутации (3, 56).
Эпидемиология
Частота диагностики LVNC у детей увеличилась за последние десятилетия, что, как считается, отражает повышение осведомленности и улучшение методов визуализации, а не рост заболеваемости (12). Данные NACCS продемонстрировали заболеваемость 0,11 на 100 000 детей в возрасте 0–10 лет и в 7 раз более высокую заболеваемость у младенцев (см. Таблицу 1). LVNC был обнаружен у 9,2% детей с диагнозом кардиомиопатия в возрасте до 10 лет (17). В ПКМР LVNC присутствовал в 4,8% случаев. Было обнаружено, что LVNC связан с дилатационным, гипертрофическим и неопределенным фенотипом в 59 случаях., 11 и 8% случаев соответственно, а изолированный НМЛЖ имел место в остальных 23% случаев. Средний возраст при постановке диагноза был значительно выше при изолированном LVNC (9,8 года) по сравнению со случаями со смешанными фенотипами (0,4–0,6 года) (12).
Особенности клинической картины
Пациенты с LVNC могут быть обнаружены при рутинном скрининге, но также могут иметь тромбоэмболические явления, аритмии или застойную сердечную недостаточность (3). Вариабельность представленных симптомов отражает фенотипическое разнообразие, и сопутствующие черты других типов кардиомиопатий вносят значительный вклад в клиническую картину. В самой большой когорте педиатрических пациентов с LVNC, из которых почти 40% были младенцами, 25% были представлены преимущественно застойной сердечной недостаточностью, 17% — аритмиями, 19% с шумом в сердце и 37% были бессимптомными (57). Положительный семейный анамнез по кардиомиопатии присутствовал в 23% всех случаев, однако только 25% этой подгруппы имели семейный анамнез по НМЛЖ.
Вариабельность представленных симптомов отражает фенотипическое разнообразие, и сопутствующие черты других типов кардиомиопатий вносят значительный вклад в клиническую картину. В самой большой когорте педиатрических пациентов с LVNC, из которых почти 40% были младенцами, 25% были представлены преимущественно застойной сердечной недостаточностью, 17% — аритмиями, 19% с шумом в сердце и 37% были бессимптомными (57). Положительный семейный анамнез по кардиомиопатии присутствовал в 23% всех случаев, однако только 25% этой подгруппы имели семейный анамнез по НМЛЖ.
Естественный анамнез
Исход педиатрической НЛЛЖ сильно варьирует и зависит от основной патофизиологии (1). Брешиа и др. рассмотрели риск смертности и внезапной смерти в крупнейшей опубликованной когорте педиатрических пациентов с LVNC. Они обнаружили сильную связь между выявленным фенотипом и риском смерти или трансплантации. Пятилетняя безтрансплантационная выживаемость была отличной для фенотипа нормального размера, промежуточной для гипертрофического фенотипа и худшей для расширенного и смешанного фенотипов (см. Таблицу 2). Наибольшими факторами риска смерти или трансплантации были наличие систолической дисфункции и/или аритмии. Внезапная сердечная смерть наступила в 6,2% случаев в течение 19 лет.-летний период исследования, при этом систолическая дисфункция присутствует у 95% и документированная аритмия у 60% этих пациентов. Раннее обращение в течение первого года жизни было дополнительным независимым фактором риска с 25% риском смерти или трансплантации при младенческом LVNC (57). Джеффрис и др. аналогичным образом наблюдали наихудший исход для пациентов с LVNC и расширенным или неопределенным фенотипом в PCMR. Риск смерти был самым высоким в первый год после установления диагноза (12). Ши и др. рассмотрели долгосрочные результаты лечения детей с LVNC из NACCS. Эта когорта включала в основном детей раннего возраста и младенцев с тяжелыми заболеваниями, средний возраст которых составлял 0,3 года на момент постановки диагноза, а застойная сердечная недостаточность присутствовала у 83% на момент постановки диагноза.
Таблицу 2). Наибольшими факторами риска смерти или трансплантации были наличие систолической дисфункции и/или аритмии. Внезапная сердечная смерть наступила в 6,2% случаев в течение 19 лет.-летний период исследования, при этом систолическая дисфункция присутствует у 95% и документированная аритмия у 60% этих пациентов. Раннее обращение в течение первого года жизни было дополнительным независимым фактором риска с 25% риском смерти или трансплантации при младенческом LVNC (57). Джеффрис и др. аналогичным образом наблюдали наихудший исход для пациентов с LVNC и расширенным или неопределенным фенотипом в PCMR. Риск смерти был самым высоким в первый год после установления диагноза (12). Ши и др. рассмотрели долгосрочные результаты лечения детей с LVNC из NACCS. Эта когорта включала в основном детей раннего возраста и младенцев с тяжелыми заболеваниями, средний возраст которых составлял 0,3 года на момент постановки диагноза, а застойная сердечная недостаточность присутствовала у 83% на момент постановки диагноза. Свобода от смерти и трансплантации составила 45% через 15 лет после постановки диагноза (см. Таблицу 1). Сопоставление показателей предрасположенности предполагает в 2 раза более высокий риск смерти и трансплантации для пациентов с LVNC и дилатационным фенотипом по сравнению с детьми с DCM из того же регистра (13). Было обнаружено, что дети с изолированным фенотипом LVNC и без наблюдаемой сердечной дисфункции имеют благоприятный исход (12). Однако в небольшой части случаев наблюдалось прогрессирование ассоциированного фенотипа кардиомиопатии с риском летального исхода, поэтому рекомендуется постоянное наблюдение (12).
Свобода от смерти и трансплантации составила 45% через 15 лет после постановки диагноза (см. Таблицу 1). Сопоставление показателей предрасположенности предполагает в 2 раза более высокий риск смерти и трансплантации для пациентов с LVNC и дилатационным фенотипом по сравнению с детьми с DCM из того же регистра (13). Было обнаружено, что дети с изолированным фенотипом LVNC и без наблюдаемой сердечной дисфункции имеют благоприятный исход (12). Однако в небольшой части случаев наблюдалось прогрессирование ассоциированного фенотипа кардиомиопатии с риском летального исхода, поэтому рекомендуется постоянное наблюдение (12).
Обсуждение
За последние два десятилетия регистры и национальные исследования предоставили важные данные об эпидемиологии и исходах детских кардиомиопатий. Зарегистрированная общая годовая заболеваемость составляла от 0,65 до 1,24 на 100 000 детей (7, 17, 18). Последовательно во всех этих эпидемиологических исследованиях ежегодная заболеваемость кардиомиопатией в течение первого года жизни была в 6-7 раз выше, чем вышеупомянутые средние показатели заболеваемости (7, 17, 18). Этот пик заболеваемости в младенчестве был обнаружен при всех типах кардиомиопатии, за исключением РКМ, которая обычно проявляется в более позднем возрасте (7, 17, 18). Данные PCMR продемонстрировали небольшой второй пик в подростковом возрасте, который был связан с ГКМП и кардиомиопатиями, вторичными по отношению к нервно-мышечным заболеваниям (7).
Этот пик заболеваемости в младенчестве был обнаружен при всех типах кардиомиопатии, за исключением РКМ, которая обычно проявляется в более позднем возрасте (7, 17, 18). Данные PCMR продемонстрировали небольшой второй пик в подростковом возрасте, который был связан с ГКМП и кардиомиопатиями, вторичными по отношению к нервно-мышечным заболеваниям (7).
Кардиомиопатия LVNC привлекает все большее внимание за последние два десятилетия, и продолжаются дискуссии относительно ее диагностических критериев (25, 26). Возрастающие показатели диагностики отражают возрастающую осведомленность об этой сущности по сравнению с предыдущими эпохами (12).
Несмотря на значительное улучшение выживаемости при врожденных пороках сердца за последние десятилетия, общие исходы детских кардиомиопатий остаются неблагоприятными (58, 59). Исключением являются выздоровление от воспалительных кардиомиопатий или успешный контроль ритма при аритмогенной сердечной недостаточности. Детские кардиомиопатии являются наиболее частым показанием к трансплантации сердца после первого года жизни (36). Самый высокий риск смерти или трансплантации сердца возникает в течение первого года после установления диагноза (19)., 22). Знание естественного течения и факторов риска неблагоприятных исходов, полученное из регистров и крупных многоцентровых исследований, помогает в стратификации риска и выборе случаев для расширенной терапии сердечной недостаточности, а также для установки ИКД для предотвращения внезапной сердечной смерти.
Самый высокий риск смерти или трансплантации сердца возникает в течение первого года после установления диагноза (19)., 22). Знание естественного течения и факторов риска неблагоприятных исходов, полученное из регистров и крупных многоцентровых исследований, помогает в стратификации риска и выборе случаев для расширенной терапии сердечной недостаточности, а также для установки ИКД для предотвращения внезапной сердечной смерти.
Заключение
Кардиомиопатии у детей, хотя и редкие, несут значительное бремя болезни из-за риска заболеваемости и смертности и отсутствия лечебной терапии. Созданные реестры предоставили ценную информацию о естественном течении и факторах риска, помогая в принятии решений по профилактике внезапной сердечной смерти и трансплантации сердца.
Авторские взносы
А.Р. разработал рукопись и разработал рисунки и таблицы. РВ руководил проектом и внес существенный вклад в окончательную версию рукописи. Оба автора внесли свой вклад в статью и одобрили представленную версию.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Примечание издателя
Все претензии, изложенные в этой статье, принадлежат исключительно авторам и не обязательно представляют претензии их дочерних организаций или издателя, редакторов и рецензентов. Любой продукт, который может быть оценен в этой статье, или претензии, которые могут быть сделаны его производителем, не гарантируются и не поддерживаются издателем.
Ссылки
1. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Детские кардиомиопатии. Цирк Рез. (2017) 121:855–73. doi: 10.1161/CIRCRESAHA.116.309386
Полный текст CrossRef | Google Scholar
2. Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Кардиомиопатия у детей: классификация и диагностика: научное заявление Американской кардиологической ассоциации. Тираж . (2019) 140:9–68. doi: 10.1161/CIR.0000000000000682
Тираж . (2019) 140:9–68. doi: 10.1161/CIR.0000000000000682
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
3. Таубин Дж. А., Лортс А., Джеффрис Дж. Л. Левожелудочковая некомпактная кардиомиопатия. Ланцет . (2015) 386:813–25. doi: 10.1016/S0140-6736(14)61282-4
CrossRef Full Text | Google Scholar
4. Weintraub RG, Semsarian C, Macdonald P. Дилатационная кардиомиопатия. Ланцет . (2017) 390:400–14. doi: 10.1016/S0140-6736(16)31713-5
CrossRef Full Text | Google Scholar
5. Денфилд С.В., Уэббер С.А. Рестриктивная кардиомиопатия в детском возрасте. Клиника сердечной недостаточности. (2010) 6:445–52. doi: 10.1016/j.hfc.2010.05.005
Полный текст CrossRef | Академия Google
6. Norrish G, Field E, McLeod K, Ilina M, Stuart G, Bhole V, et al. Клиническая картина и выживаемость гипертрофической кардиомиопатии у детей: ретроспективное исследование в Соединенном Королевстве. Евро Сердце J .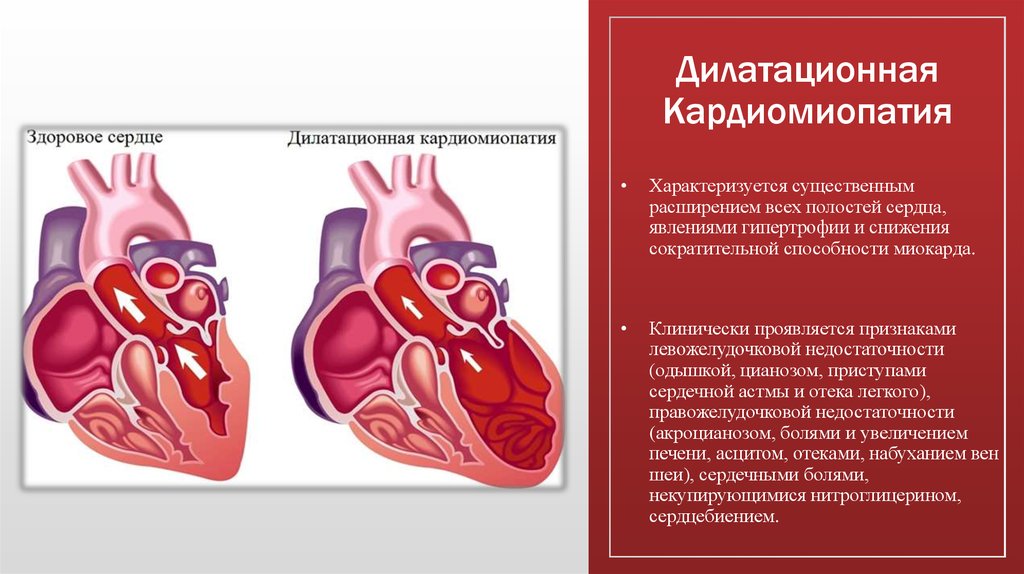 (2019) 40:986–93. doi: 10.1093/eurheartj/ehy798
(2019) 40:986–93. doi: 10.1093/eurheartj/ehy798
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
7. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. Заболеваемость детской кардиомиопатией в двух регионах США. N Английский J Med . (2003) 348:1647–55. doi: 10.1056/NEJMoa021715
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
8. Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, et al. Отдаленные результаты гипертрофической кардиомиопатии, диагностированной в детстве: результаты национального популяционного исследования. Тираж . (2018) 138:29–36. doi: 10.1161/CIRCULATIONAHA.117.028895
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
9. Maurizi N, Passantino S, Spaziani G, Girolami F, Arretini A, Targetti M, et al. Отдаленные исходы детской гипертрофической кардиомиопатии и возрастные факторы риска летальных аритмий. JAMA Кардиол . (2018) 3: 520–5. doi: 10.1001/jamacardio.2018.0789
(2018) 3: 520–5. doi: 10.1001/jamacardio.2018.0789
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
10. Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD, Addonizio LJ, et al. Исходы рестриктивной кардиомиопатии в детстве и влияние фенотипа: отчет из регистра детской кардиомиопатии. Тираж . (2012) 126:1237–44. doi: 10.1161/CIRCULATIONAHA.112.104638
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
11. Weller RJ, Weintraub R, Addonizio LJ, Chrisant MRK, Gersony WM, Hsu DT. Исходы идиопатической рестриктивной кардиомиопатии у детей. Ам Дж Кардиол . (2002) 90:501–6. doi: 10.1016/S0002-9149(02)02522-5
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
12. Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al. Фенотипы и исходы кардиомиопатии у детей с некомпактным миокардом левого желудочка: результаты регистра детской кардиомиопатии. Ошибка карты J .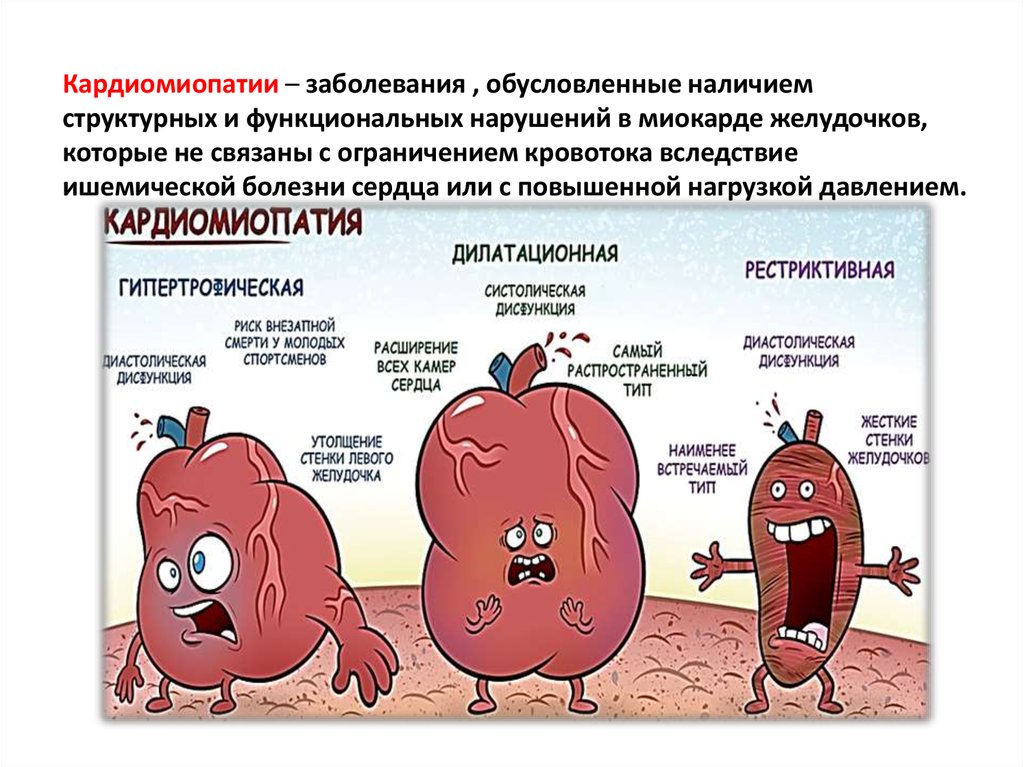 (2015) 21:877–84. doi: 10.1016/j.cardfail.2015.06.381
(2015) 21:877–84. doi: 10.1016/j.cardfail.2015.06.381
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
13. Shi WY, Betancur MM, Nugent AW, Cheung M, Colan S, Turner C, et al. Отдаленные исходы некомпактной кардиомиопатии левого желудочка у детей: результаты национального популяционного исследования. Тираж . (2018) 138:367–76. doi: 10.1161/CIRCULATIONAHA.117.032262
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
14. Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE. Регистр детской кардиомиопатии: 1995-2007 гг. Прог Педиатр Кардиол . (2008) 25:31–6. doi: 10.1016/j.ppedcard.2007.11.006
CrossRef Full Text | Google Scholar
15. Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, et al. Регистр детской кардиомиопатии и сердечной недостаточности: основные результаты первых 15 лет. Клиника сердечной недостаточности . (2010) 6:401–13. doi: 10.1016/j.hfc. 2010.05.002
2010.05.002
Реферат PubMed | Полный текст перекрестной ссылки | Google Scholar
16. Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, et al. Естественное течение дилатационной кардиомиопатии у детей. J Am Heart Assoc . (2016) 5:1–10. doi: 10.1161/JAHA.116.003450
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
17. Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. Эпидемиология детской кардиомиопатии в Австралии. N Английский J Med . (2003) 348:1639–46. doi: 10.1056/NEJMoa021737
CrossRef Полный текст | Google Scholar
18. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, et al. Эпидемиология идиопатических кардиомиопатий у детей и подростков: общенациональное исследование в Финляндии. Am J Эпидемиол . (1997) 146:385–93. doi: 10.1093/oxfordjournals.aje.a009291
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
19. Эндрюс Р.Е., Фентон М.Дж., Ридаут Д.А., Берч М. Впервые возникшая сердечная недостаточность из-за заболевания сердечной мышцы в детстве: проспективное исследование в Соединенном Королевстве и Ирландии. Тираж . (2008) 117:79–84. doi: 10.1161/CIRCULATIONAHA.106.671735
Эндрюс Р.Е., Фентон М.Дж., Ридаут Д.А., Берч М. Впервые возникшая сердечная недостаточность из-за заболевания сердечной мышцы в детстве: проспективное исследование в Соединенном Королевстве и Ирландии. Тираж . (2008) 117:79–84. doi: 10.1161/CIRCULATIONAHA.106.671735
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
20. Таубин Дж. А., Лоу А. М., Колан С. Д., Слипер Л. А., Орав Э. Дж., Клуни С. и другие. Заболеваемость, причины и исход дилатационной кардиомиопатии у детей. JAMA Кардиол. (2006) 296:1867–76. doi: 10.1001/jama.296.15.1867
Полный текст CrossRef | Google Scholar
21. Daubeney PEF, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Клинические особенности и исходы дилатационной кардиомиопатии у детей: результаты национального популяционного исследования. Тираж . (2006) 114:2671–8. doi: 10.1161/CIRCULATIONAHA.106.635128
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
22. Alexander PMA, Daubeney PEF, Nugent AW, Lee KJ, Turner C, Colan SD, et al. Отдаленные результаты дилатационной кардиомиопатии, диагностированной в детстве: результаты национального популяционного исследования детской кардиомиопатии. Тираж . (2013) 128:2039–46. doi: 10.1161/CIRCULATIONAHA.113.002767
Alexander PMA, Daubeney PEF, Nugent AW, Lee KJ, Turner C, Colan SD, et al. Отдаленные результаты дилатационной кардиомиопатии, диагностированной в детстве: результаты национального популяционного исследования детской кардиомиопатии. Тираж . (2013) 128:2039–46. doi: 10.1161/CIRCULATIONAHA.113.002767
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
23. Колан С.Д., Липшульц С.Е., Лоу А.М., Слипер Л.А., Мессере Дж., Кокс Г.Ф. и соавт. Эпидемиология и причинно-специфический исход гипертрофической кардиомиопатии у детей: данные регистра детской кардиомиопатии. Тираж . (2007) 115:773–81. doi: 10.1161/CIRCULATIONAHA.106.621185
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
24. Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Клинические особенности и исходы детской гипертрофической кардиомиопатии: результаты национального популяционного исследования. Тираж . (2005) 112:1332–8.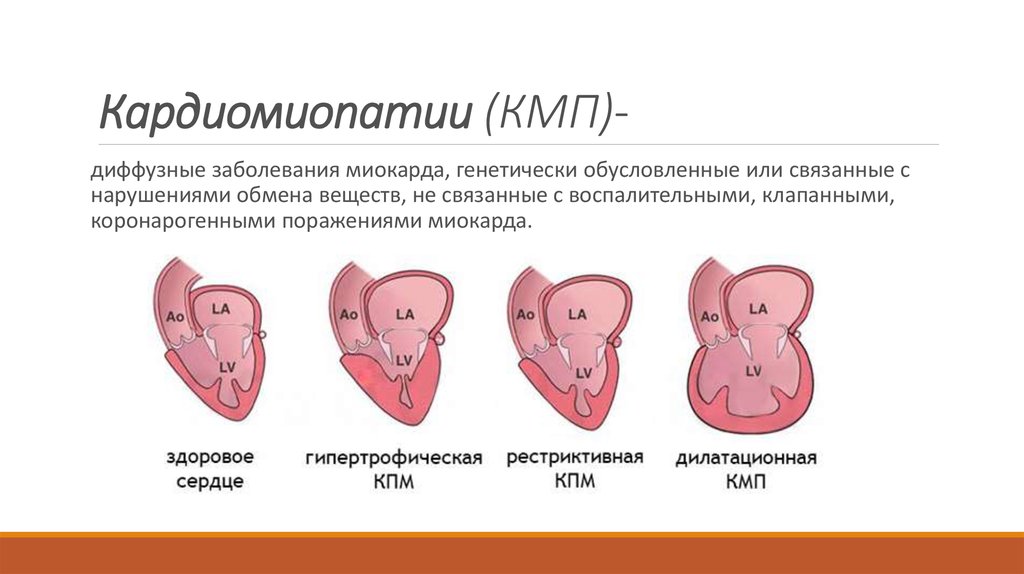 doi: 10.1161/CIRCULATIONAHA.104.530303
doi: 10.1161/CIRCULATIONAHA.104.530303
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
25. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Классификация кардиомиопатий: заявление рабочей группы Европейского общества кардиологов по миокардиальным и перикардиальным заболеваниям. Eur Heart J. (2008) 29: 270–6. doi: 10.1093/eurheartj/ehm342
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
26. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Современные определения и классификация кардиомиопатий: научное заявление Американской кардиологической ассоциации Совета по клинической кардиологии, сердечной недостаточности и комитету по трансплантации; исследования и функции качества медицинской помощи и результатов. Тираж . (2006) 113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287
CrossRef Полный текст | Google Scholar
27. Konta L, Franklin RCG, Kaski JP. Номенклатура и системы классификации кардиомиопатий у детей. Кардиол Янг . (2015) 25:31–42. doi: 10.1017/S1047951115001201
Номенклатура и системы классификации кардиомиопатий у детей. Кардиол Янг . (2015) 25:31–42. doi: 10.1017/S1047951115001201
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
28. McNally EM, Mestroni L. Дилатационная кардиомиопатия: генетические детерминанты и механизмы. Circ Res . (2017) 121:731–48. doi: 10.1161/CIRCRESAHA.116.309396
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
29. Bondue A, Arbustini E, Bianco A, Ciccarelli M, Dawson D, De Rosa M, et al. Сложные пути от генотипа к фенотипу при дилатационной кардиомиопатии: обновленная научная информация от рабочей группы функции миокарда Европейского общества кардиологов. Кардиоваскулярный рез . (2018) 114:1287–303. doi: 10.1093/cvr/cvy122
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
30. Nugent AW, Davis AM, Kleinert S, Wilkinson JL, Weintraub RG. Клинические, электрокардиографические и гистологические корреляции у детей с дилатационной кардиомиопатией. J Трансплантация сердца и легкого . (2001) 20:1152–7. doi: 10.1016/S1053-2498(01)00334-5
J Трансплантация сердца и легкого . (2001) 20:1152–7. doi: 10.1016/S1053-2498(01)00334-5
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
31. Белкая С., Конторович А.Р., Бьюн М., Мулеро-Наварро С., Баджолле Ф., Кобат А. и др. Аутосомно-рецессивная кардиомиопатия, проявляющаяся острым миокардитом. J Am Coll Кардиол. (2017) 69:1653–65. doi: 10.1016/j.jacc.2017.01.043
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
32. Campuzano O, Fernández-Falgueras A, Sarquella-Brugada G, Sanchez O, Cesar S, Mademont I, et al. Генетически уязвимый миокард может предрасполагать к миокардиту. J Am Coll Cardiol . (2015) 66:2913–4. doi: 10.1016/j.jacc.2015.10.049
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
33. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. Роль эндомиокардиальной биопсии в лечении сердечно-сосудистых заболеваний: научное заявление Американской кардиологической ассоциации, Американского колледжа кардиологов и Европейского общества кардиологов, одобренное Американским обществом сердечной недостаточности и Ассоциацией сердечной недостаточности Европейского общества кардиологии.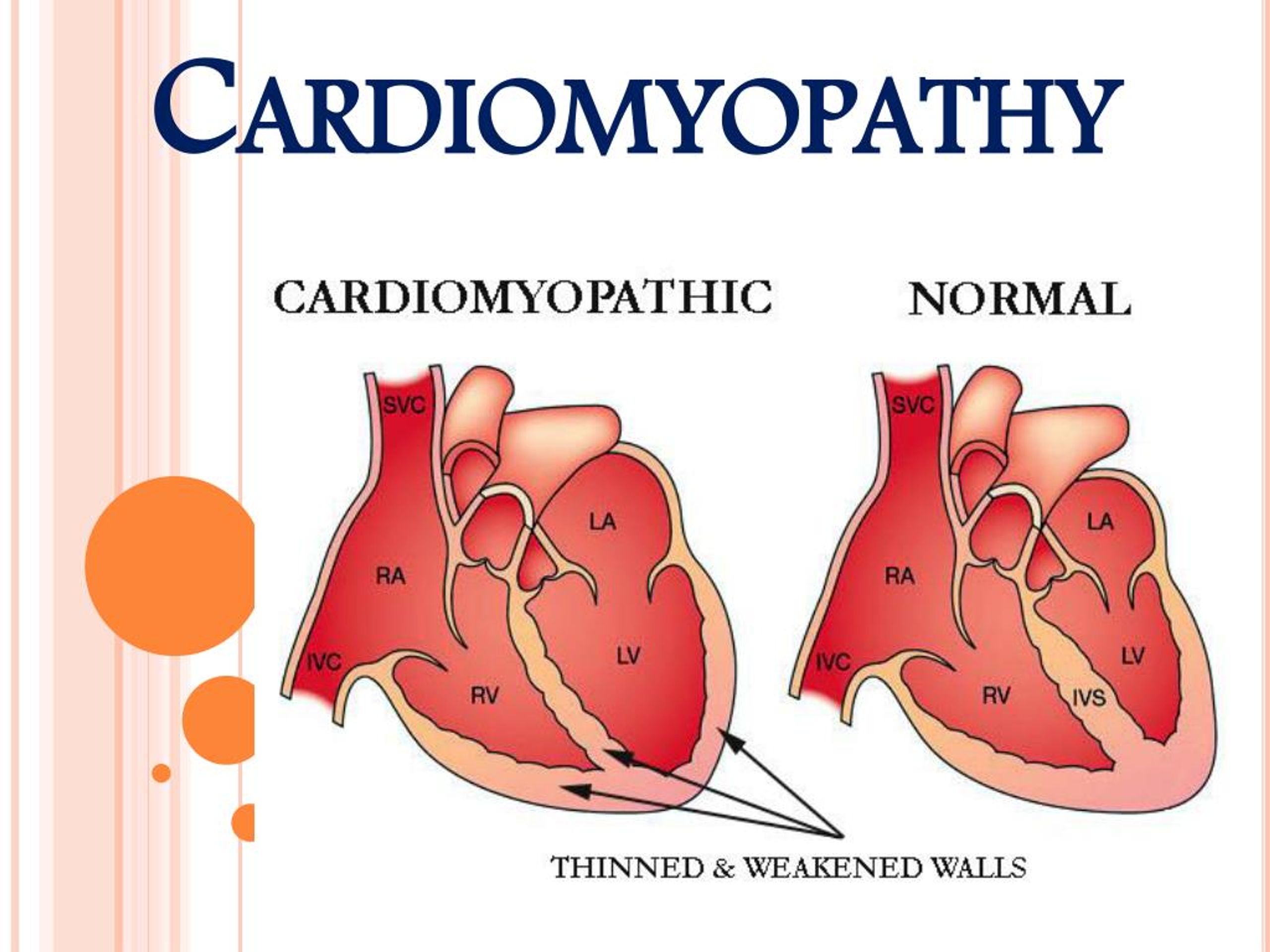 Eur Heart J. (2007) 28:3076–93. doi: 10.1093/eurheartj/ehm456
Eur Heart J. (2007) 28:3076–93. doi: 10.1093/eurheartj/ehm456
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
34. Canter CE, Simpson KP. Диагностика и лечение миокардита у детей в современное время. Тираж . (2014) 129:115–28. doi: 10.1161/CIRCULATIONAHA.113.001372
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
35. Shamszad P, Hall M, Rossano JW, Denfield SW, Knudson JD, Penny DJ, et al. Особенности и исходы госпитализации детей с кардиомиопатией в связи с сердечной недостаточностью. Ошибка карты J . (2013) 19:672–7. doi: 10.1016/j.cardfail.2013.08.006
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
36. Хсу Д.Т., Ламур Дж.М., Кантер К.Э. Заболевания сердца, приводящие к педиатрической трансплантации: кардиомиопатии и врожденные пороки сердца. Педиатрическая трансплантация сердца. ISHLT Monogr Series 2. Addison, TX: Int Soc Heart Lung Transp. (2008) 2.
37. Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, et al. Восстановление эхокардиографической функции у детей с идиопатической дилатационной кардиомиопатией: результаты регистра детской кардиомиопатии. J Am Coll Cardiol . (2014) 63:1405–13. doi: 10.1016/j.jacc.2013.11.059
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
38. den Boer SL, Rizopoulos D, du Marchie Sarvaas GJ, Backx APCM, ten Harkel ADJ, van Iperen GG, et al. Полезность серийных измерений N-концевого про-В-типа натрийуретического пептида для прогнозирования сердечной смерти при острой и хронической дилатационной кардиомиопатии у детей. Ам Дж Кардиол . (2016) 118:1723–9. doi: 10.1016/j.amjcard.2016.08.053
Реферат PubMed | Полный текст перекрестной ссылки | Google Scholar
39. Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, Webber SA, et al. Частота и факторы риска внезапной сердечной смерти у детей с дилатационной кардиомиопатией: отчет из реестра детской кардиомиопатии. J Am Coll Cardiol . (2012) 59:607–15. doi: 10.1016/j.jacc.2011.10.878
J Am Coll Cardiol . (2012) 59:607–15. doi: 10.1016/j.jacc.2011.10.878
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
40. Singh RK, Canter CE, Shi L, Colan SD, Dodd DA, Everitt MD, et al. Выживаемость без трансплантации сердца у детей с дилатационной кардиомиопатией. J Am Coll Cardiol . (2017) 70:2663–73. doi: 10.1016/j.jacc.2017.09.1089
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
41. Марстон Н.А., Хан Л., Оливотто И., Дэй С.М., Эшли Э.А., Михелс М. и др. Клинические характеристики и исходы гипертрофической кардиомиопатии в детском возрасте. Евро Сердце J . (2021) 42:1988–96. doi: 10.1093/eurheartj/ehab148
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
42. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, et al. Исходы у детей с синдромом Нунан и гипертрофической кардиомиопатией: исследование из реестра детской кардиомиопатии. Ам Сердце J . (2012) 164:442–8. doi: 10.1016/j.ahj.2012.04.018
(2012) 164:442–8. doi: 10.1016/j.ahj.2012.04.018
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
43. Norrish G, Cantarutti N, Pissaridou E, Ridout DA, Limongelli G, Elliott PM, et al. Факторы риска внезапной сердечной смерти при детской гипертрофической кардиомиопатии: систематический обзор и метаанализ. Eur J Prev Cardiol . (2017) 24:1220–30. doi: 10.1177/2047487317702519
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
44. Norrish G, Ding T, Field E, Ziółkowska L, Olivotto I, Limongelli G, et al. Разработка новой модели прогнозирования риска внезапной сердечной смерти при детской гипертрофической кардиомиопатии (дети с риском ГКМП). JAMA Кардиол . (2019) 4: 918–27. doi: 10.1001/jamacardio.2019.2861
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
45. Мирон А., Лафреньер-Рула М., Стив Фан С.П., Армстронг К.Р., Драгулеску А., Папаз Т. и др. Утвержденная модель прогнозирования риска внезапной сердечной смерти при детской гипертрофической кардиомиопатии. Тираж . (2020) 142: 217–29. doi: 10.1161/CIRCULATIONAHA.120.047235
Тираж . (2020) 142: 217–29. doi: 10.1161/CIRCULATIONAHA.120.047235
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
46. Balaji S, DiLorenzo MP, Fish FA, Etheridge SP, Aziz PF, Russell MW, et al. Факторы риска летальных аритмических событий у детей и подростков с гипертрофической кардиомиопатией и имплантируемым дефибриллятором: международное многоцентровое исследование. Услышать ритм . (2019) 16:1462–7. doi: 10.1016/j.hrthm.2019.04.040
Реферат PubMed | Полный текст перекрестной ссылки | Google Scholar
47. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, et al. Эпидемиология смерти, связанной с гипертрофической кардиомиопатией. Тираж . (2000) 102:858–64. doi: 10.1161/01.CIR.102.8.858
CrossRef Полный текст | Google Scholar
48. Kaski JP, Syrris P, Burch M, Tomé Esteban MT, Fenton M, Christiansen M, et al. Идиопатическая рестриктивная кардиомиопатия у детей вызывается мутациями в генах белков кардиального саркомера. Сердце . (2008) 94:1478–84. doi: 10.1136/hrt.2007.134684
Сердце . (2008) 94:1478–84. doi: 10.1136/hrt.2007.134684
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
49. Wittekind SG, Ryan TD, Gao Z, Zafar F, Czosek RJ, Chin CW, et al. Современные исходы детской рестриктивной кардиомиопатии: одноцентровый опыт. Педиатр Кардиол. (2019) 40: 694–704. doi: 10.1007/s00246-018-2043-0
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
50. Кушваха С.С., Фэллон Дж.Т., Фустер В. Рестриктивная кардиомиопатия. N Английский J Med . (1997) 336: 267–76. doi: 10.1056/NEJM199701233360407
CrossRef Full Text | Google Scholar
51. Ривенс С.М., Кирни Д.Л., Смит Э.ОБ., Таубин Дж.А., Денфилд С.В. Внезапная смерть и сердечно-сосудистый коллапс у детей с рестриктивной кардиомиопатией. Тираж . (2000) 102:876–82. doi: 10.1161/01.CIR.102.8.876
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
52. Уолш М.А., Гренье М. А., Джеффрис Дж.Л., Таубин Дж.А., Лортс А., Чосек Р.Дж. Нарушения проводимости у детей с рестриктивной кардиомиопатией. Сердечная недостаточность . (2012) 5: 267–73. doi: 10.1161/CIRCHEARTFAILURE.111.964395
А., Джеффрис Дж.Л., Таубин Дж.А., Лортс А., Чосек Р.Дж. Нарушения проводимости у детей с рестриктивной кардиомиопатией. Сердечная недостаточность . (2012) 5: 267–73. doi: 10.1161/CIRCHEARTFAILURE.111.964395
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
53. Руссо Л.М., Уэббер С.А. Идиопатическая рестриктивная кардиомиопатия у детей. Сердце. (2005) 91:1199–202. doi: 10.1136/hrt.2004.043869
CrossRef Full Text | Google Scholar
54. Андерсон Х.Н., Сетта Ф., Дрисколл Д.Дж., Олсон Т.М., Акерман М.Дж., Джонсон Дж.Н. Идиопатическая рестриктивная кардиомиопатия у детей и молодых людей. Ам Дж Кардиол . (2018) 121:1266–70. doi: 10.1016/j.amjcard.2018.01.045
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
55. Weir-McCall JR, Yeap PM, Papagiorcopulo C, Fitzgerald K, Gandy SJ, Lambert M, et al. Неуплотнение левого желудочка: анатомический фенотип или отчетливая кардиомиопатия? J Am Coll Cardiol . (2016) 68:2157–65. doi: 10.1016/j.jacc.2016.08.054
(2016) 68:2157–65. doi: 10.1016/j.jacc.2016.08.054
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
56. van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, et al. Генетика, клинические особенности и отдаленные результаты некомпактной кардиомиопатии. J Am Coll Cardiol . (2018) 71:711–22. doi: 10.1016/j.jacc.2017.12.019
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
57. Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Смертность и внезапная смерть у детей с некомпрессией левого желудочка в третичном специализированном центре. Тираж . (2013) 127:2202–8. doi: 10.1161/CIRCULATIONAHA.113.002511
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
58. McKenna WJ, Maron BJ, Thiene G. Классификация, эпидемиология и глобальное бремя кардиомиопатий. Circ Res . (2017) 121:722–30. doi: 10.1161/CIRCRESAHA. 117.309711
117.309711
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
59. Zimmerman MS, Smith AGC, Sable CA, Echko MM, Wilner LB, Olsen HE, et al. Глобальное, региональное и национальное бремя врожденных пороков сердца, 1990–2017 гг.: систематический анализ исследования глобального бремени болезней, 2017 г. Ланцет для детского лечения подростков . (2020) 4: 185–200. doi: 10.1016/S2352-4642(19)30402-X
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Кардиомиопатия | Cincinnati Children’s
Кардиомиопатии — это заболевания сердечной мышцы, также известные как миокард, при которых поражаются собственно мышечные клетки и окружающие ткани.
Кардиомиопатии могут быть первичными, то есть болезнь поражает сердце.
Они также могут быть вторичными, то есть болезнь является результатом другого заболевания или токсина, а также может поражать другие органы тела, включая сердце.
У пациентов с кардиомиопатией чаще всего сердце выглядит нормальным, но плохо работает.
Причины кардиомиопатии
Причина кардиомиопатии в большинстве случаев неизвестна, ее также называют идиопатической. Некоторые причины кардиомиопатии могут включать:
- Изменения в генетическом коде или ДНК, которые могут передаваться по наследству
- Вирусная инфекция сердца или миокардит, который ослабляет сердечную мышцу
- Нарушение обмена веществ, а также наследственные заболевания мышц
- Проблемы с коронарными артериями, имеющиеся при рождении или приобретенные
Хотя существует длинный список возможных причин кардиомиопатии, немногие из них поддаются непосредственному лечению, и большая часть терапии направлена на устранение вторичных эффектов на сердце.
Существуют и другие менее распространенные формы кардиомиопатии, которые могут быть вызваны дефицитом гормонов, хроническими заболеваниями и нарушениями сердечного ритма. В рамках оценки кардиомиопатии проводится подробный медицинский и семейный анамнез, чтобы найти причину кардиомиопатии.
Симптомы кардиомиопатии
У детей кардиомиопатия может проявляться по-разному. У некоторых детей нет никаких симптомов, и их просят обратиться к кардиологу, потому что в их семье есть сердечные заболевания, их врач замечает изменения в их медицинском осмотре или из-за отклонений от нормы, которые были сделаны по другой причине. У ребенка также могут быть признаки и симптомы застойной сердечной недостаточности (ЗСН). В некоторых очень редких случаях внезапное сердечное событие, связанное с аномальным сердечным ритмом, может быть первым признаком проблемы с сердцем у ребенка.
У младенцев с кардиомиопатией могут быть:
- Учащенное и тяжелое дыхание в покое или во время кормления
- Кормление длится дольше, чем обычно
- Потоотделение при кормлении
- Повышенная утомляемость или малоподвижность
- Плохая прибавка в весе
Общие симптомы у детей старшего возраста и подростков:
- Боль в животе
- Тошнота/рвота
- Снижение аппетита
- Боль в груди
- Проблемы с физическими упражнениями или неспособность не отставать от других детей своего возраста
- Проблемы с дыханием
- Кашель
- Отек лица, живота или ног
- Аномальные сердечные сокращения или учащенное сердцебиение
- Головокружение
- Потеря сознания (потеря сознания во время физических упражнений является серьезной проблемой, поэтому детей следует осмотреть кардиолог)
Диагностика кардиомиопатии
Физикальное обследование
Для всех детей с подозрением на кардиомиопатию сбор анамнеза и физикальное обследование являются ключевыми в постановке диагноза. Признаки застойной сердечной недостаточности, такие как учащенное дыхание и учащенное сердцебиение, аномальные легочные и сердечные тоны и увеличение печени, могут помочь врачу поставить правильный диагноз.
Признаки застойной сердечной недостаточности, такие как учащенное дыхание и учащенное сердцебиение, аномальные легочные и сердечные тоны и увеличение печени, могут помочь врачу поставить правильный диагноз.
Шум или патологическая импульсация грудной стенки могут быть обнаружены при кардиомиопатии, но их отсутствие не исключает диагноз.
Шум или аномальные импульсы грудной стенки могут быть обнаружены при кардиомиопатии. При некоторых типах кардиомиопатии шум может быть связан с обструкцией кровотока, выходящего из сердца, или несостоятельностью одного из клапанов сердца. Если у ребенка нет шума, это не означает, что у него нет кардиомиопатии. Родителей, братьев и сестер ребенка с кардиомиопатией также могут попросить посетить кардиолога, даже если у них нет признаков или симптомов.
Тест для диагностики Включите:
- Электрокардиограмму (ЭКГ) для проверки сердечного ритма.
- Эхокардиограмма — это УЗИ сердца. Этот тест оценивает размер и форму сердца, а также то, насколько хорошо сердце сжимает/качает кровь.
- Магнитно-резонансная томография (МРТ) — это еще один способ оценить размер, форму и функцию сердца, и ее можно проводить вместо эхокардиограммы. Иногда вводят внутривенное (в/в) контрастное вещество, чтобы лучше рассмотреть саму сердечную мышцу.
- Рентгенологическое исследование органов грудной клетки иногда используется для скрининга сердечных заболеваний и может показать увеличенное сердце и избыточную жидкость в легких (также называемую отеком легких).
- Сердечно-легочная проба с нагрузкой проводится на беговой дорожке или велосипеде для детей старшего возраста и подростков. Это помогает оценить, насколько хорошо ребенок может тренироваться, и посмотреть на сердечный ритм, частоту сердечных сокращений и артериальное давление во время упражнений.
- Холтеровское мониторирование или монитор событий — это другие тесты для мониторинга сердечного ритма. Их носят на груди от 24 часов до 30 дней и помогают выявить нарушения сердечного ритма.
- Также может быть выполнена катетеризация сердца не только для оценки давления в каждой камере сердца, но и для оценки коронарных артерий. Иногда при катетеризации сердца также берут небольшие кусочки сердечной мышцы для лабораторного исследования – это называется биопсией. Такие биопсии сердечной мышцы полезны при оценке возможных инфекций сердца, а также некоторых метаболических нарушений сердца.
Типы кардиомиопатии
Кардиологи разделяют кардиомиопатию на пять категорий:
- Дилатационная кардиомиопатия (ДКМП) , также известная как застойная кардиомиопатия: Дилатационная кардиомиопатия является наиболее распространенной формой кардиомиопатии у детей, а ишемическая кардиомиопатия является наиболее распространенным типом дилатационной кардиомиопатии. При ДКМП сердце увеличивается в размерах и плохо работает.
- Гипертрофическая кардиомиопатия (ГКМП) : При гипертрофической кардиомиопатии сердечная мышца утолщается.
 Другое его название — идиопатический гипертрофический субаортальный стеноз (IHSS). Утолщение сердечной мышцы, особенно стенок левой половины сердца, может затруднять ток крови через сердце и его полное расслабление. Дети с гипертрофической кардиомиопатией также подвержены повышенному риску нарушения сердечного ритма
Другое его название — идиопатический гипертрофический субаортальный стеноз (IHSS). Утолщение сердечной мышцы, особенно стенок левой половины сердца, может затруднять ток крови через сердце и его полное расслабление. Дети с гипертрофической кардиомиопатией также подвержены повышенному риску нарушения сердечного ритма - Рестриктивная кардиомиопатия (РКМ) : При рестриктивной кардиомиопатии насосная камера или желудочек не могут хорошо расслабиться. В результате кровь возвращается в верхние камеры сердца или предсердия, и они увеличиваются или увеличиваются в размерах, в то время как нижние насосные камеры, желудочки, остаются нормальными по размеру. Существует также риск нарушения сердечного ритма.
- Левый желудочек без уплотнения (LVNC): На эхокардиограмме сердечная мышца выглядит грубой и отмечена пальцевидными выступами, называемыми трабекулами. Он кажется грубым и зубчатым, а не гладким и компактным. Это может быть связано с другими формами врожденных пороков сердца, но может возникать и само по себе.
 У некоторых пациентов может развиться плохая функция и сердечная недостаточность.
У некоторых пациентов может развиться плохая функция и сердечная недостаточность. - Аритмогенная дисплазия правого желудочка (АДПЖ): Мышца сердца со временем замещается жировой тканью и приводит к опасным сердечным ритмам. Эту форму кардиомиопатии часто трудно диагностировать, и она может поражать правую и/или левую сторону сердца. Обычно это впервые обнаруживается, когда у пациента наблюдается ненормальный ритм. Это также иногда называют аритмогенной кардиомиопатией правого желудочка (ARVC) или аритмогенной дисплазией правого желудочка (ARVD).
Лечение кардиомиопатии
Лечение детей с кардиомиопатией лучше всего разделить на неотложные и долгосрочные временные рамки.
Неотложная помощь
Если ребенок находится в критическом состоянии, для лечения могут потребоваться меры по спасению жизни, такие как установка дыхательной трубки и использование механического вентилятора. Пациентам с острым заболеванием могут потребоваться внутривенные жидкости и лекарства для улучшения артериального давления, функции сердца и сердечного ритма.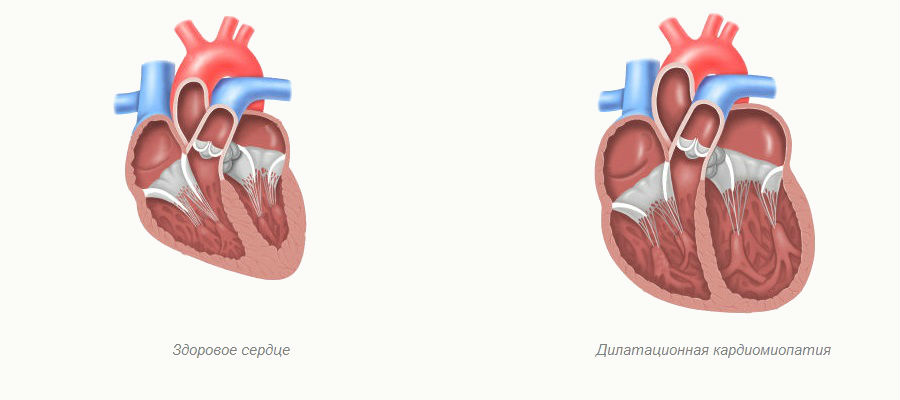 Если ребенок очень болен, ему может понадобиться помощь аппарата искусственного кровообращения, называемого ЭКМО. Вспомогательное устройство для желудочков также можно использовать, если функция сердца сохраняется.
Если ребенок очень болен, ему может понадобиться помощь аппарата искусственного кровообращения, называемого ЭКМО. Вспомогательное устройство для желудочков также можно использовать, если функция сердца сохраняется.
У пациентов с опасным для жизни сердечным ритмом, связанным с кардиомиопатией, хирургически размещенные устройства могут мгновенно «электризовать» пациента во время опасного для жизни события. Такие устройства называются ИКД (внутренний кардиовертер/дефибриллятор).
Долгосрочный уход
Когда состояние ребенка станет более стабильным, долгосрочный уход может включать ряд стратегий, включая использование лекарств, частые посещения и тестирование. В зависимости от типа кардиомиопатии могут использоваться различные виды лекарств. Лекарства от кровяного давления, такие как ингибиторы АПФ (ACEi) и бета-блокаторы, часто используются, если сердце не сжимается, как в случае с ДКМП. Диуретики, такие как Лазикс, — это лекарства, предназначенные для удаления лишней жидкости из организма. Разбавители крови используются, когда в сердце образуются сгустки.
Разбавители крови используются, когда в сердце образуются сгустки.
Оценка риска аномального сердечного ритма у ребенка, связанного с кардиомиопатией и ИКД, может быть плановой процедурой.
Гипертрофическая кардиомиопатия
Неотложная помощь
Пациенты с гипертрофической кардиомиопатией могут иметь проблемы с аномальной, учащенной частотой сердечных сокращений. Им могут потребоваться лекарства для замедления частоты сердечных сокращений (бета-блокаторы) или даже электрошок, чтобы остановить эти аномальные ритмы. Эти пациенты также могут нуждаться в тех же спасательных процедурах, упомянутых выше.
Длительное лечение
Более хронические варианты лечения гипертрофической кардиомиопатии направлены как на проблемы с сердечным ритмом, так и на проблемы с обструкцией кровотока. Лечение обструкции оттока может включать такие лекарства, как бета-блокаторы (атенолол, метопролол) и блокаторы кальциевых каналов (верапамил), предназначенные для замедления частоты сердечных сокращений и «расслабления» сердца, тем самым уменьшая обструкцию.
Рестриктивная кардиомиопатия
Неотложная и долгосрочная помощь
Пациенты с рестриктивной кардиомиопатией подвержены высокому риску образования тромбов в сердце, особенно в расширенных верхних камерах. Могут потребоваться препараты для разжижения крови, такие как аспирин, кумадин (варфарин) или ловенокс. Некоторым пациентам также может помочь умеренное использование диуретиков. Внутренние кардиовертер-дефибрилляторы (ИКД) также используются, потому что у этих пациентов существует риск внезапной остановки сердца, связанной с быстрым или медленным ритмом.
АРВД
Неотложная и долгосрочная помощь<
Пациентам с АКМ может помочь лечение лекарственными препаратами для ограничения нарушений ритма. Лечение также заключается в размещении ИКД для защиты от внезапных сердечных событий, таких как остановка сердца. В некоторых случаях может быть выполнена катетерная процедура для устранения аномального ритма.
Долгосрочные перспективы для детей с кардиомиопатией
Дилатационная кардиомиопатия (ДКМП) — серьезное заболевание. Однако, как и большинство заболеваний, дилатационная кардиомиопатия протекает с различной степенью тяжести и исходами.
Однако, как и большинство заболеваний, дилатационная кардиомиопатия протекает с различной степенью тяжести и исходами.
Среди пациентов с DCM и сердечной недостаточностью, вызванной вирусными заболеваниями или миокардитом, примерно у одной трети наблюдается стойкое нарушение функции сердца, у одной трети состояние улучшается, но остается некоторая сердечная дисфункция, а у одной трети полностью выздоравливает. Трудно предсказать, в какую категорию попадет конкретный пациент. Вот почему так важно частое наблюдение у кардиолога. Пациентам с необратимыми повреждениями и стойкими нарушениями функции сердца может потребоваться пересадка сердца.
Точное количество пациентов с гипертрофической кардиомиопатией неизвестно, так как у некоторых пациентов симптомы отсутствуют. Вероятность преждевременной смерти оценивается менее чем в 1 процент. Факторы риска внезапной смерти включают эпизоды обморока, диагноз в молодом возрасте, семейный анамнез внезапной смерти, выраженное утолщение сердца на эхокардиограмме и учащенный сердечный ритм, наблюдаемый при мониторировании. Менее чем у 5% пациентов поздние осложнения могут включать увеличение левого желудочка и снижение насосной функции.
Менее чем у 5% пациентов поздние осложнения могут включать увеличение левого желудочка и снижение насосной функции.
Поскольку рестриктивная кардиомиопатия приходится только на 5 процентов пациентов с кардиомиопатией, общее число пациентов с этим заболеванием невелико, а данные об общих исходах ограничены. К сожалению, имеющаяся информация указывает на неблагоприятный прогноз. По оценкам, только от 45 до 50 процентов пациентов с этим типом кардиомиопатии выживают через два года после постановки диагноза. Этих пациентов обычно направляют на трансплантацию сердца на ранней стадии, до того, как у них появятся другие симптомы болезни сердца.
Неуплотнение левого желудочка может проявляться по-разному, и у некоторых пациентов никогда не развивается болезнь сердца. Семейный анамнез в этих случаях важен для выявления пациентов с риском развития сердечно-сосудистых заболеваний, но большинство пациентов находятся в изоляции. Необходимы регулярные визиты к кардиологу для обнаружения каких-либо изменений в работе сердца.
Исход АРВД трудно оценить из-за его редкого характера и поздней диагностики. Первоначальное состояние пациента, например, после остановки сердца или семейного анамнеза, может повлиять на течение болезни.
О кардиомиопатии у детей | Детское здоровье ColumbiaDoctors
Записаться на прием
Наша команда готова помочь вам записаться на прием к нужным вам специалистам.
Факты, которые нужно знать
- Кардиомиопатия поражает примерно одного из каждых 100 000 детей в США в возрасте до 18 лет.
- Начало кардиомиопатии чаще встречается у младенцев в возрасте до 12 месяцев по сравнению с детьми более старшего возраста.
- Наиболее распространенной формой кардиомиопатии у детей является дилатационная кардиомиопатия.
Что такое кардиомиопатия у детей?
Термин кардиомиопатия используется для описания редкой группы заболеваний, поражающих сердечную мышцу. Сердечная мышца может стать толстой, увеличенной, тонкой или жесткой, и болезнь может поразить верхние камеры (предсердия), нижние камеры (желудочки) или и то, и другое. Кардиомиопатия нарушает способность сердца правильно перекачивать кровь или поддерживать нормальный ритм.
Сердечная мышца может стать толстой, увеличенной, тонкой или жесткой, и болезнь может поразить верхние камеры (предсердия), нижние камеры (желудочки) или и то, и другое. Кардиомиопатия нарушает способность сердца правильно перекачивать кровь или поддерживать нормальный ритм.
У детей кардиомиопатия может иметь неизвестную причину (идиопатическая кардиомиопатия), и иногда ее диагностируют пренатально. Заболевание может быть генетическим, что означает, что генная мутация передается из поколения в поколение. Это также может быть результатом инфекции (миокардит) или редко быть вызвано более генерализованным заболеванием (например, метаболическим синдромом, мышечной дистрофией или заболеванием коллагеновых сосудов). Наконец, токсические агенты, такие как химиотерапевтические препараты, могут быть еще одной идентифицируемой причиной кардиомиопатии у детей и подростков.
Существует четыре типа кардиомиопатии. Каждый вид требует разного лечения.
- Дилатационная кардиомиопатия: Наиболее распространенный тип кардиомиопатии, который возникает, когда желудочки — основные насосные камеры сердца — увеличиваются и растягиваются (расширяются).
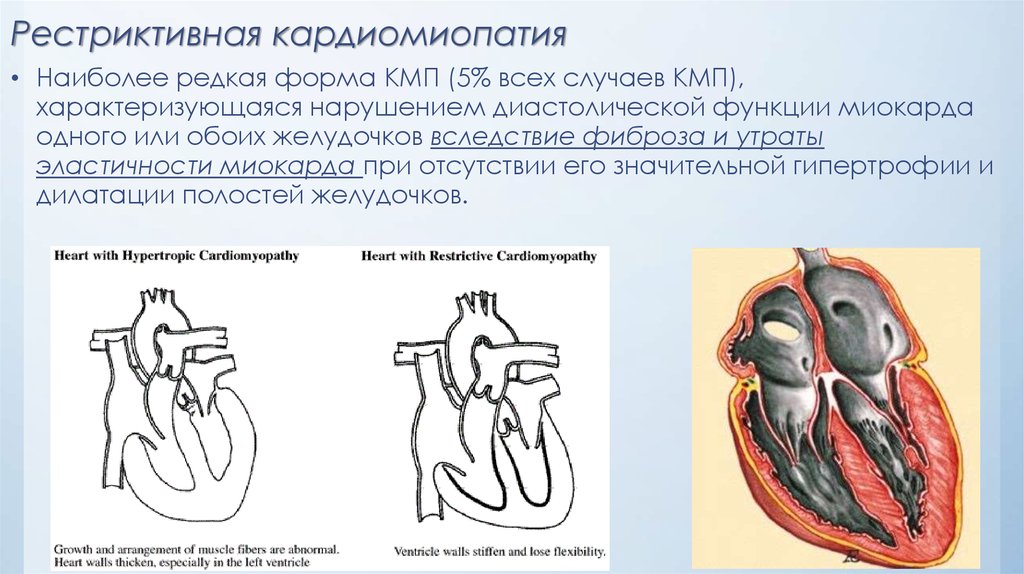 На ранних стадиях у детей симптомы могут отсутствовать, но по мере прогрессирования заболевания развиваются симптомы сердечной недостаточности.
На ранних стадиях у детей симптомы могут отсутствовать, но по мере прогрессирования заболевания развиваются симптомы сердечной недостаточности. - Гипертрофическая кардиомиопатия: Утолщение мышцы между двумя желудочками и/или стенками одного или обоих желудочков. Гипертрофическая кардиомиопатия может проявляться опасным для жизни нарушением сердечного ритма или проявляться сердечной недостаточностью или болью в груди.
- Некомпрессионная кардиомиопатия: Форма кардиомиопатии с различными симптомами, при которой сердечная мышца очень аномально сформирована. Сердце может стать растянутым, как при дилатационной кардиомиопатии, или ригидным, как при гипертрофической кардиомиопатии. Дети могут протекать бессимптомно или могут быть диагностированы в очень молодом возрасте.
- Рестриктивная кардиомиопатия: Редкий тип кардиомиопатии, при котором сердечная мышца становится жесткой, что затрудняет правильное наполнение камер кровью.
 Детям с этим типом кардиомиопатии может потребоваться обследование для пересадки сердца вскоре после постановки диагноза.
Детям с этим типом кардиомиопатии может потребоваться обследование для пересадки сердца вскоре после постановки диагноза.
Каковы симптомы кардиомиопатии?
Иногда у детей при рождении проявляются симптомы кардиомиопатии. У других детей симптомы отсутствуют, пока они не станут старше. Симптомы кардиомиопатии у новорожденных включают:
- Учащенное или затрудненное дыхание
- Учащенное сердцебиение
- Проблемы с кормлением
- Чрезмерная потливость
- Плохая прибавка в весе
- Раздражительность или отсутствие реакции
- Необычный цвет кожи
Симптомы у детей старшего возраста включают:
- Аномальное сердцебиение
- Одышка при физической нагрузке или снижение толерантности к физической нагрузке
- Проблемы с желудком: периодическая рвота, тошнота, снижение аппетита, неспособность принимать пищу
- Боль в груди
- Головокружение или обморок
- Необычная усталость
- Учащенное сердцебиение
- Вздутие живота или вздутие живота
- Необычный цвет кожи
Как диагностируется кардиомиопатия?
Для диагностики кардиомиопатии у вашего ребенка мы, скорее всего, проведем ряд исследований. К ним могут относиться:
К ним могут относиться:
- Рентген грудной клетки: может показать увеличение сердца.
- Электрокардиограмма (ЭКГ или ЭКГ): записывает электрическую активность сердца для выявления нарушений ритма.
- Эхокардиография (эхо): оценивает структуру и функцию сердца с помощью ультразвуковых волн.
- Магнитно-резонансная томография (МРТ): неинвазивный инструмент визуализации, который показывает подробные изображения сердца, точно измеряет размеры камер и может выявить рубцевание сердечной мышцы.
- Катетеризация сердца: минимально инвазивная процедура, при которой длинная тонкая трубка, называемая катетером, вводится в сердце через кровеносные сосуды через крошечный разрез в паху или на шее. Катетер используется для осмотра сердца, измерения давления в сердечных камерах и легочных сосудах и, возможно, для получения биопсии (образца) сердечной ткани. В определенных ситуациях требуется биопсия, после чего крошечный кусочек сердечной мышцы можно исследовать в лаборатории, что может помочь в диагностике нескольких конкретных типов кардиомиопатии.
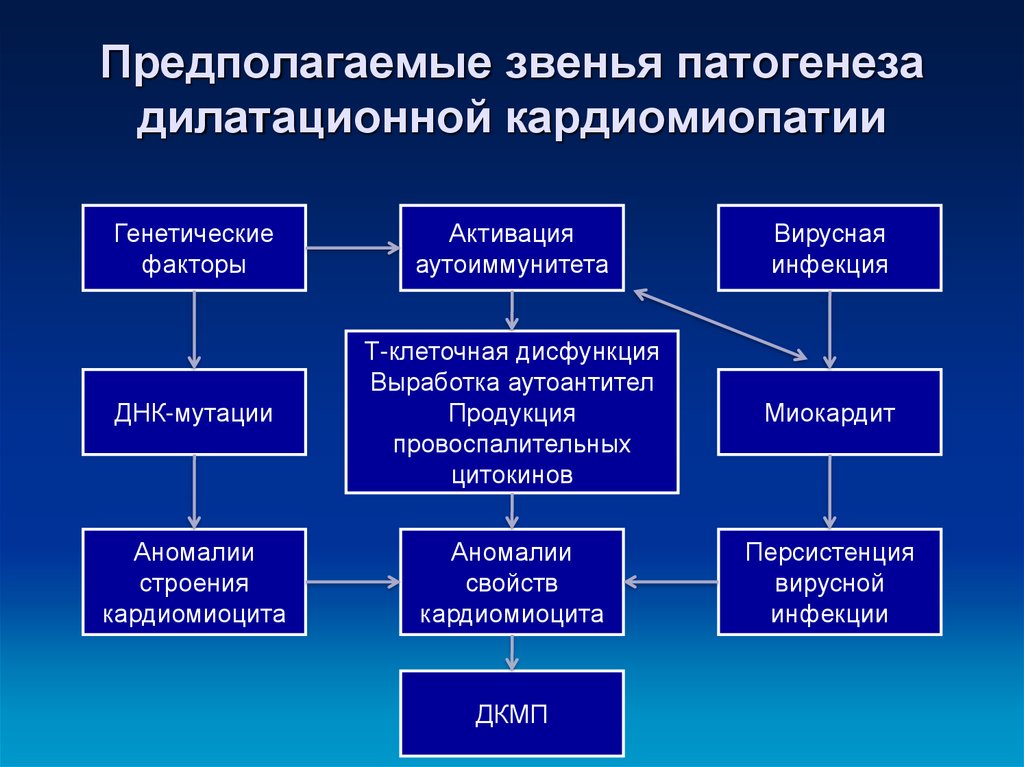
- Радионуклидное тестирование: использует крошечные количества низкодозированных радиоактивных материалов, называемых трассерами, которые вводятся в вену и светятся под специальными камерами по мере продвижения к сердцу. Камеры могут создавать изображения сердца на основе высвобождаемой энергии и могут отслеживать аномальный приток крови к сердечной мышце и более конкретные признаки того, как сердце работает в условиях стресса.
- Генетическое тестирование: ищет генные мутации, связанные с кардиомиопатией.
Как лечится кардиомиопатия в Колумбийском университете?
Эксперты Программы детской кардиомиопатии, сердечной недостаточности и трансплантологии подбирают лечение в зависимости от типа кардиомиопатии и болезни сердца, от которых страдает пациент, и тяжести каждого случая. Когда кардиомиопатия имеет генетическую причину, мы оказываем помощь всем членам семьи в нашей Детской интегрированной клинике кардиогенетики.