
Эпидермолиз врожденный: Congenital epidermolysis bullosa: modern methods of diagnosis and therapy. Prospects for regenerative medicine — Kubanov

Врожденный эпидермолиз Текст научной статьи по специальности «Клиническая медицина»
Проблемы здоровья и экологии
89
СЛУЧАЙ ИЗ КЛИНИЧЕСКОЙ ПРАКТИКИ
УДК 616.5-002.157-053.1-052.31
ВРОЖДЕННЫЙ ЭПИДЕРМОЛИЗ
О. А. Румянцева, А. И. Зарянкина, Л. В. Кривицкая,
Т. Е. Бубневич, И. М. Малолетникова
Гомельский государственный медицинский университет
Буллезный эпидермолиз (epidermolysis bullosa) — один из самых тяжелых наследственных дерматозов. Особенность и сложность данной патологии связаны не только с физическим дискомфортом пациентов и тяжестью заболевания, что часто приводит к инвалидизации, но и с развитием особых фенотипических проявлений, которые приводят к снижению качества жизни и нарушению социально-психологической адаптации.
На сегодняшний день методов этиопатогенетического лечения врожденного эпидермолиза не разработано. Все существующие методы лечения являются симптоматическими и направлены в основном на уход за пациентом.
Ключевые слова: врожденный эпидермолиз, новорожденный, лечение, уход.
CONGENITAL EPIDERMOLYSIS
О. А. Rumyantseva, А. I. Zaryankina, L. V. Krivitskaya,
T. Е. Bubnevich, I. M. Maloletnikova
Gomel State Medical University
Еpidermolysis bullosa is one of the most severe hereditary dermatoses. Peculiarities and complexity of the pathology are associated not only with the physical discomfort of patients and the severity of the disease, which often causes disability, but also with the development of specific phenotypic manifestations leading to reduced quality of life and worsening of social and psychological adaptation.
By now no ethiopathogenetic treatment methods to cure hereditary epidermolysis bullosa have been found. All available methods of treatment are symptomatic and are mainly aimed at patient care.
Key words: epidermolysis bullosa, newborn, treatment, care.
Буллезный эпидермолиз (БЭ) — гетерогенная группа заболеваний, развивающихся вследствие врожденного дефекта в генах, кодирующих разные протеины дермоэпидермального соединения. Проявляется образованием пузырей на коже и слизистых оболочках, подвергающихся трению. В зависимости от уровня поражения включает в себя несколько типов.
Проявляется образованием пузырей на коже и слизистых оболочках, подвергающихся трению. В зависимости от уровня поражения включает в себя несколько типов.
Термин «буллезный эпидермолиз» (БЭ) впервые был использован в 1886 г. Кебнером, хотя случаи, подобные данному диагнозу, описывались и ранее. В конце XIX — начале XX века Брок и Аллоп (Brocq and Hallopeau) ввели термины: «травматическая пузырчатка» (Traumatic pemphigus), «врожденное травматическое образование пузырей» (congenital traumatic blistering) и «буллезный акантолизм» (acantholysis bullosa), которые на сегодняшний день вышли из употребления [1, 2]. В 1962 г. Пирсоном была разработана первая классификация, основанная на применении трансмиссионной электронной микроскопии. Были выделены три основных типа БЭ, основанных на данных об ультраструктурном уровне образования пузы-
рей у пациентов с БЭ: эпидермолитический (простой), люцидолитический (пограничный) и дермолитический (дистрофический). В 1980-х годах были использованы поликлональные и моноклональные антитела при исследовании кожи при БЭ, была разработана техника иммунофлюоресцентного антигенного картирования. Почти для каждого общепринятого подтипа БЭ был определен молекулярный дефект в генах, кодирующих структурные белки кожи человека. За последние годы знания о БЭ значительно расширились как на клиническом, так и на молекулярном уровнях. В 2007 г. состоялся последний пересмотр классификации БЭ, которая была опубликована в 2008 г. [1, 2].
Почти для каждого общепринятого подтипа БЭ был определен молекулярный дефект в генах, кодирующих структурные белки кожи человека. За последние годы знания о БЭ значительно расширились как на клиническом, так и на молекулярном уровнях. В 2007 г. состоялся последний пересмотр классификации БЭ, которая была опубликована в 2008 г. [1, 2].
Средняя распространенность буллезного эпидермолиза в мире 1,7 : 100 тыс. населения. Один из 227 человек имеет мутацию в гене, отвечающем за развитие буллезного эпидермолиза. Оба пола поражаются одинаково.
В зависимости от уровня отслойки эпидермиса от дермы выделяют четыре основных типа БЭ: простой, пограничный, дистрофический и синдром Киндлера [3, 4].
Проблемы здоровья и экологии
90
Основной чертой простого БЭ являются интраэпидермальные пузыри. Это — аутосом-но-доминантный тип наследования. Простой БЭ обусловлен дефектом гена кератина К5 или К14. Он существует в трех основных формах: Вебера-Коккейна, Кебнера, Даулинга-Мира.
У пациентов с пограничным БЭ пузыри формируются в светлой пластинке, у пациентов с дистрофическим — под плотной пластинкой базальной мембраны кожи («дермоэпидермальное соединение»).
Пограничный БЭ наследуется аутосомно-рецессивно. Выделяют четыре формы:
1. Пограничный буллезный эпидермолиз Херлитца (дефект ламинина 5). Высыпания обычно присутствуют с рождения или возникают сразу после него, пузыри образуются на любой части тела, включая слизистые оболочки, характерно разрастание грануляционной ткани вокруг рта, может поражаться гортань, часто наблюдается дистрофия ногтей или ано-нихия, может осложниться сепсисом, вследствие чего возрастает процент летальности.
2. Пограничный буллезный эпидермолиз с атрезией привратника (дефект интегрина аб/р4). Возможно сочетание атрезии привратника и пороков развития мочеполовой системы, прогноз неблагоприятный.
3. Пограничный буллезный эпидермолиз, отличный от формы Херлитца (дефект коллагена XVII). Такие же проявления, как и при пограничном БЭ Херлитца, но в более легкой форме, слизистые оболочки поражены в меньшей степени.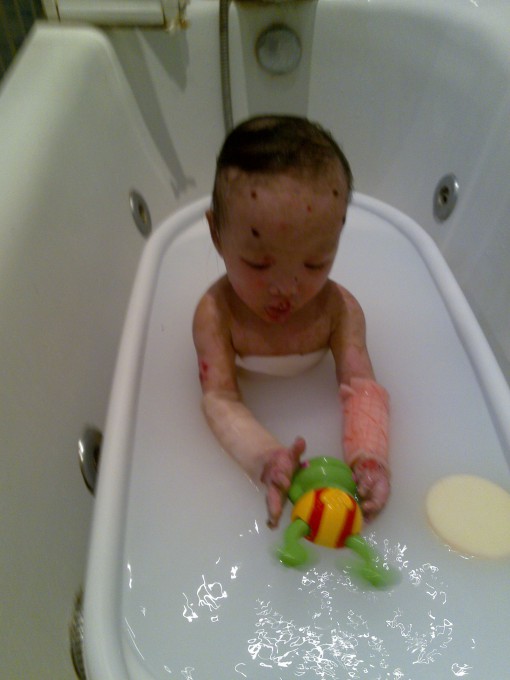
4. Пограничный буллезный эпидермолиз с мышечной дистрофией (дефект плектина). Напоминает БЭ легкой степени тяжести, развивается в любое время, начиная с периода новорожденности и заканчивая третьим десятилетием жизни.
Дистрофический тип БЭ (дефект коллагена VII) может наследоваться доминантно и рецессивно. При доминантном дистрофическом буллезном эпидермолизе формирование пузырей наиболее выражено в дистальных отделах конечностей, на локтях и коленях. Характерны милиумы, на месте пузырей формируются рубцы, характерна дистрофия ногтей. При рецессивном дистрофическом БЭ пузыри присутствуют с рождения, поражена кожа и слизистые оболочки, формируется большое количество рубцов, наблюдается деформация кистей и стоп по типу варежек и носков со сращением пальцев между собой, часто наблюдается кариес зубов, возможна задержка прорезывания зубов. Из-за рубцевания кожи вокруг рта формируется микростомия. Среди других осложнений наблюдаются нарушение глотания
(вследствие рубцевания пищевода), хроническая анемия, задержка роста, формирование рубцов на конъюнктиве, предрасположенность к развитию плоскоклеточной карциномы [2, 3].
При синдроме Киндлера множественные участки разрушения могут быть видны на разном уровне кожи. Возникают распространенные пузырные образования, формирующиеся к моменту рождения, позже характерны светочувствительность и пойкилодермия, дистрофия ногтевых пластинок и атрофические рубцы. Внекожные проявления включают острый колит, эзофагит, уретральные стриктуры, реже — выворот века. Зубы не подвергаются патологическим изменениям, но возможно развитие гингивальной гиперплазии [3].
Представляется описание случая у ребенка с диагнозом: «Дерматолитический тип врожденного буллезного эпидермолиза не-Hallopo-Simense, дистрофический, тяжелое течение. Врожденные перетяжки нижней трети правой голени, правого предплечья. Задержка моторного развития. Анемия полифакторная, умеренной степени тяжести. Контрактуры первых пальцев стоп, кистей. Множественные эрозии слизистой оболочки полости рта, языка (эпи-телизированы). Малая аномалия сердца: аномальные диагональные трабекулы левого желудочка. НК0».
НК0».
Ребенок Т. (мальчик) от четвертой беременности, четвертых срочных родов в 39-40 недель. Беременность протекала на фоне кольпита, угрозы прерывания в 12, 23, 30-31 неделе, истмико-цервикальной недостаточности, гестационного пиелонефрита, ферментопатии, ОРИ — в 37 недель. Родился с массой тела 3590 г. в тяжелом состоянии за счет интоксикации на фоне обширных дефектов кожных покровов, в большей степени на конечностях. Оба родителя здоровы. В роддоме выхаживался в условиях реанимационного отделения. На 4-е сутки жизни был переведен в отделение анестезиологии и реанимации УЗ «Гомельская областная детская клиническая больница» в тяжелом состоянии за счет болевого, интоксикационного синдромов на фоне массивного поражения кожных покровов. Отмечалась умеренная кислородозависимость (FiO2 21 %). Крик громкий, болезненный. На коже головы, туловища, конечностей — пузыри со светлым и геморрагическим содержимым, кровоточат на конечностях, эрозии по типу «обожженной кожи», некротические изменения на стопах, атрофические изменения кожи на правой голени, амниотические перетяжки правого голеностопного сустава, правого предплечья. Поражение кожных покровов составляло более 25 % (рисунки 1, 2).
Поражение кожных покровов составляло более 25 % (рисунки 1, 2).
Проблемы здоровья и экологии
91
Рисунки 1, 2 — Пузыри, эрозии по типу «обожженной кожи», некротические изменения на стопах
В связи с поражением слизистых оболочек ротовой полости энтеральное питание было затруднено, болезненно, вызывало образование пузырей и эрозивных поверхностей. Получал питание через зонд, эпизодически — через рожок. Дыхание ритмичное, периодически — диспноэ. При аускультации дыхание равномерное, жесткое. ЧД до 62 в минуту. Сердечные тоны ритмичные, приглушены, тахикардия до 188/мин (на фоне болевого синдрома при перевязках, купании ребенка).
Ребенок находился в оАиР в течение 3 месяцев. На всем протяжении лечения отмечалось волнообразное течение: мнимое благополучие сменялось ухудшением (ежедневно-генерализованное образование пузырей с различным содержимым,
после дренажа которых появлялись эрозии с дальнейшим образованием некроза тканей, в большей степени на стопах, кистях). При кормлении из рожка образовывались пузыри, которые спонтанно вскрывались с образованием эрозий в ротовой полости, отмечалась дисфагия. На фоне массивного поражения кожных покровов и слизистых наблюдалась гипопротеинемия (общий белок снижался до 42 г/л на фоне подачи белка 10 г/кг/сут), анемия тяжелой степени, что требовало трансфузии трижды отмытых эритроцитов. Присоединение вторичной флоры ухудшало течение заболевания. В динамике развились атрофические рубцы, олигодистрофия с последующей утратой ногтей, с формированием контрактур первых пальцев кистей, стоп (рисунки 3, 4).
При кормлении из рожка образовывались пузыри, которые спонтанно вскрывались с образованием эрозий в ротовой полости, отмечалась дисфагия. На фоне массивного поражения кожных покровов и слизистых наблюдалась гипопротеинемия (общий белок снижался до 42 г/л на фоне подачи белка 10 г/кг/сут), анемия тяжелой степени, что требовало трансфузии трижды отмытых эритроцитов. Присоединение вторичной флоры ухудшало течение заболевания. В динамике развились атрофические рубцы, олигодистрофия с последующей утратой ногтей, с формированием контрактур первых пальцев кистей, стоп (рисунки 3, 4).
Рисунок 3 — Олигодистрофия Рисунок 4 — Атрофические рубцы запястья
В диагностике проводилось исключение внутриутробных инфекций методом ИФА и ПЦР, политопный микробиологический и вирусологический контроль биологических сред, в том числе из раневой поверхности, контроль показателей общего анализа крови, мочи, биохимического анализа крови, инструментальные исследования: рентгенологическое исследование органов грудной клетки, ультразвуковое исследование голов-
ного мозга, сердца, органов брюшной полости и забрюшинного пространства. Неоднократно проводились консилиумы с участием офтальмолога, комбустиолога, дерматолога, генетика, ортопеда, невролога, ЛОР-врача, стоматолога.
Неоднократно проводились консилиумы с участием офтальмолога, комбустиолога, дерматолога, генетика, ортопеда, невролога, ЛОР-врача, стоматолога.
В лечении использовались антибактериальные препараты, начиная с защищенных пе-нициллинов и до назначения препаратов резерва в связи с присоединением вторичной флоры, ин-
Проблемы здоровья и экологии
92
фузионная терапия с целью возмещения потерь тканевой жидкости, дезинтоксикационная терапия. Огромная роль уделялась энтеральному питанию с высокой дотацией белка и каллоража адаптированными смесями с использованием обогатителей БМ-85 (у матери отмечалась гипога-лактия), при ухудшении состоянии — дотация белка парентерально (ваминолакт, альбумин), которая суммарно составляла 10 г/кг/сут. В процессе эпителизации раневой поверхности дотация белка уменьшалась.
Кормление осуществлялось из бутылочки с клапаном с использованием «супермягких» силиконовых сосок, а также из силиконовой ложечки.
Проводилась трансфузия трижды отмытых эритроцитов с целью купирования тяжелой анемии, в последующем — применение препаратов железа, токоферола, витамина Д, ретинола ацетата.
Для обезболивания использовались препараты из группы нестероидных противовоспалительных средств.
Осуществлялся тщательный уход за кожными покровами и слизистыми оболочками: слизистые ротовой полости обрабатывались облепиховым маслом, десна перед каждым кормлением — дентиноксом с целью обезболивания, для носовых ходов и глаз применялись капли Бепантен, проводился немедленный дренаж в асептических условиях образовавшихся пузырей без деформирования кожи, купание в смягчающих средствах для атопичной кожи фирмы
«Avenue» с последующим использованием дезинфицирующих кремов (Бепантен плюс, Судокрем, Дермазин для атопичной кожи «Avenue», при необходимости — Октенисепт). Для ухода за ранами пациента использовались Протонсан раствор и гель, атравматичные салфетки Мedicomp от Paul Hartman, для «засохших» поверхностей — спрей Нилтан. С целью уменьшения количества перевязок, а значит, снижения болевого синдрома применялись первичные и вторичные повязки: Урго-тюль, Мепитель — для свежих ран, Мепилекс обычный, «Трансфер», «Лайт», Аутраман, Бранолид, Гидротюль, Д-4. С целью защиты уязвимых мест проводилось бинтование мягкими бинтами ОАО «Лента» «Доктор Бинт» (Могилев), Пеха-креп, фиксация Пеха-хавт от Paul Hartman, а также использовались штюль-пы или трубчатые эластичные бинты. С целью контроля наличия стула применялся прелакс.
С целью защиты уязвимых мест проводилось бинтование мягкими бинтами ОАО «Лента» «Доктор Бинт» (Могилев), Пеха-креп, фиксация Пеха-хавт от Paul Hartman, а также использовались штюль-пы или трубчатые эластичные бинты. С целью контроля наличия стула применялся прелакс.
Мама была обучена особенностям кормления ребенка с БЭ, ухода за кожными покровами, купания, навыкам наложения повязок и бинтования.
Благодаря тщательному уходу за кожными покровами, использованию первичных и вторичных повязок дефекты кожи эпителизи-ровались, буллезные высыпания стали значительно реже, в основном в местах сгибов (рисунки 5, 6). Однако сформировались атрофические рубцы, олигодистрофия, утрата ногтей на всех пальчиках, а также контрактуры пальцев кистей, стоп.
Рисунки 5, 6 — Единичные пузыри/эрозивные поверхности перед выпиской
Выводы
Лечение детей с БЭ остается паллиативным, направленным на уход за эрозивноязвенными поражениями кожи, на раннее выявление и лечение осложнений.
Мультидисциплинарное сотрудничество врачей, обеспечение пациентам оптимального ухода и лечения, обучение родителей правильному уходу за ребенком, позволяют достичь длительной ремиссии, а значит, улучшить качество его жизни, уменьшить инвалидизирующие последствия.
библиографический список
1. Детская дерматология: справочник / под ред. Д. П. Кро-учука, А. Дж. Манчини; пер. с англ.; под ред. Н. Г. Короткого. — М.: Практическая медицина, 2010. — 608 с.
2. Европейское руководство по лечению дерматологичских болезней / под ред. А. Д. Кацамбаса, Т. М. Лотти. — М.: МЕД прессинформ, 2008. — С. 94-103.
3. Аяьбанова, В. И. Буллезный эпидермолиз: первый год жизни / В. И. Альбанова // Рос вестник перинатологии и педиатрии. — 2010. — № 3. — С. 110-117.
4. Кожные и венерические болезни. Руководство / под ред. Ю. К. Скрипкина, В. Н. Мордовцева. — М : Медицина, 1999. — С. 789-813.
Поступила 08.05.2015
Врождённый буллёзный эпидермолиз | Городской кожно-венерологический диспансер
В Городском центре дерматологии и венерологии создан кабинет врождённого буллёзного эпидермолиза (ВБЭ) — генетически обусловленного заболевания, характеризующегося спонтанным появлением пузырей, эрозий, возможным вовлечением в процесс всего кожного покрова и развитием тяжелых осложнений.
Врач-дерматовенеролог кабинета ВБЭ осуществляет:
- ведение учета больных с ВБЭ;
- оказание квалифицированной медицинской помощи, в том числе на дому;
- наблюдение за пациентами с ВБЭ с целью выявления и профилактики возможных осложнений;
- оказание паллиативной терапии.
Запись к врачу-дерматовенерологу кабинета ВБЭ осуществляется по направлению врачей-дерматовенерологов Санкт-Петербурга по предварительной записи:
- по телефону: (812)246-38-49, (812)246-38-68;
- при личном обращении в регистратуру ГЦДВ (запись ведётся ежедневно, кроме выходных и праздничных дней, с 9.00 до 14.00).
Для приёма врача-дерматовенеролога кабинета ВБЭ пациенту необходимо предъявить в регистратуре:
- направление на консультацию от врача-дерматовенеролога районного КВД;
- паспорт РФ или свидетельство о рождении ребенка;
- действующий полис ОМС;
- СНИЛС (страховой номер индивидуального лицевого счёта в системе обязательного пенсионного страхования РФ).

В СПб ГБУЗ «ГорКВД» создана врачебная комиссия по работе с пациентами с ВБЭ с целью принятия решения о предоставлении медицинских изделий для проведения паллиативной терапии. Организация оказания паллиативной терапии пациентам с ВБЭ проводится за счёт средств бюджета города. Запись на врачебную комиссию осуществляет врач-дерматовенеролог кабинета ВБЭ.
Со списком необходимых документов для получения медицинских изделий для проведения паллиативной терапии пациентам, страдающим ВБЭ, можно ознакомиться ЗДЕСЬ.
Порядок принятия решения об обеспечении и выдачи медицинских изделий определены постановлением Правительства Санкт-Петербурга и распоряжением Комитета по здравоохранению.
Школьникова Татьяна Валерьевна
Школьникова Татьяна Валерьевна
к.м.н., врач-дерматовенеролог высшей квалификационной категории, заведующая ГЦДВ
Закон Санкт-Петербурга от 09.
 11.2011 № 728-132 «Социальный кодекс Санкт-Петербурга»
11.2011 № 728-132 «Социальный кодекс Санкт-Петербурга»
Городской центр дерматологии и венерологии (ГЦДВ) СПб ГБУЗ «ГорКВД»
Адрес: 192102, г. Санкт-Петербург, наб. реки Волковки, д. 3, литер А (2 этаж)
Телефон: 8 (812) 246-38-49, 8 (812) 246-38-60 (доб. 3)
СПб ГБУЗ «Городской кожно-
венерологический диспансер»
192102, г. Санкт-Петербург,
наб. реки Волковки, д. 3
Противодействие коррупции
Доступная среда
Страховые медицинские организации
Взаимодействие с СОНКО
Сайт разработан bistem.ru — Enfold Theme by Kriesi
Буллезный эпидермолиз — NHS
Буллезный эпидермолиз (БЭ) — это название группы редких наследственных кожных заболеваний, при которых кожа становится очень хрупкой. Любая травма или трение кожи может вызвать болезненные волдыри.
Симптомы буллезного эпидермолиза
Общие симптомы при всех типах БЭ включают:
- кожа, которая легко покрывается волдырями
- волдыри на руках и подошвах ног
- утолщение кожи, которое со временем может покрыться рубцами или изменить цвет
- утолщение кожи и ногтей
Кредит:
НАУЧНАЯ ФОТОБИБЛИОТЕКА https://www. sciencephoto.com/media/151100/view
sciencephoto.com/media/151100/view
Кредит:
НАУЧНАЯ ФОТОБИБЛИОТЕКА https://www.sciencephoto.com/media/877183/view
Типы буллезного эпидермолиза
3 основных типа ЭБ:
- Простой буллезный эпидермолиз (EBS) – наиболее распространенный тип, который может варьироваться от легкого, с низким риском серьезных осложнений, до тяжелого
- дистрофический буллезный эпидермолиз (DEB) – который может варьироваться от легкого до тяжелого
- пограничный буллезный эпидермолиз (ПБЭ) – редкая форма БЭ, варьирующая от умеренной до тяжелой
Тип отражает, где на теле образуются волдыри и какой слой кожи поражен.
Существует также много вариантов этих 3 основных типов БЭ, каждый из которых имеет несколько разные симптомы.
Узнайте больше о симптомах различных типов буллезного эпидермолиза.
Диагностика ЭБ
БЭ обычно диагностируется у младенцев и детей младшего возраста, поскольку симптомы могут быть очевидны с самого рождения. Но некоторые более легкие формы БЭ могут быть не диагностированы до наступления совершеннолетия.
Если есть подозрение, что у вашего ребенка заболевание, его направят к дерматологу (дерматологу).
Специалист проведет анализы для определения типа БЭ и поможет составить план лечения. Они могут взять небольшой образец кожи (биопсия) для отправки на анализ.
Пренатальное тестирование
В некоторых случаях можно проверить неродившегося ребенка на БЭ после 11-й недели беременности.
Пренатальные тесты включают амниоцентез и биопсию ворсин хориона.
Это может быть предложено, если известно, что вы или ваш партнер являетесь носителем дефектного гена, связанного с БЭ, и существует риск рождения ребенка с тяжелой формой БЭ.
Если тест подтвердит, что у вашего ребенка будет БЭ, вам предложат консультацию и совет.
Причины буллезного эпидермолиза
БЭ вызывается неисправным геном (генная мутация), который делает кожу более хрупкой.
Ребенок с БЭ мог унаследовать дефектный ген от родителя, у которого также есть БЭ. Или они могли унаследовать дефектный ген от обоих родителей, которые являются просто «носителями», но сами не имеют БЭ.
Изменение гена также может произойти случайно, если ни один из родителей не является носителем.
Лечение буллезного эпидермолиза
В настоящее время нет лекарства от БЭ, поэтому лечение направлено на облегчение симптомов и предотвращение развития осложнений, таких как инфекция.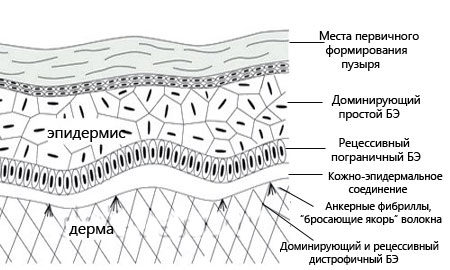
Группа врачей-специалистов поможет вам решить, какое лечение лучше всего подходит для вашего ребенка, и посоветует, как жить с этим заболеванием.
Лечение БЭ может включать:
- выдавливание волдырей стерильной иглой
- наложение защитных повязок
- избегание вещей, которые ухудшают состояние
Лекарства можно использовать для лечения инфекции или уменьшения боли. Хирургическое вмешательство может потребоваться, если БЭ вызывает сужение пищевода или проблемы с руками.
Узнайте больше о лечении буллезного эпидермолиза.
Приобретенный буллезный эпидермолиз
Приобретенный буллезный эпидермолиз (ПБЭ) представляет собой приобретенную форму БЭ со сходными симптомами.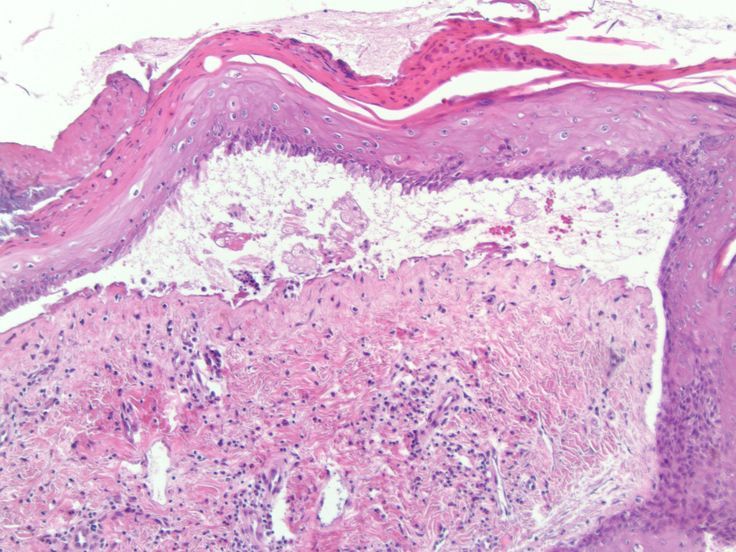
Как и при БЭ, при ЭБК на коже легко образуются волдыри. Он также может поражать полость рта, горло и желудочно-кишечный тракт.
Но EBA не передается по наследству, и симптомы обычно не проявляются до более позднего возраста.
Это аутоиммунное заболевание, означающее, что ваша иммунная система начинает атаковать здоровые ткани организма. Точно неизвестно, чем это вызвано.
ЭБА — это очень редкое заболевание, которым чаще всего страдают люди старше 40 лет.
Благотворительные организации и группы поддержки
Если у вашего ребенка диагностирован БЭ, это может быть пугающим и ошеломляющим опытом. Вероятно, вы захотите узнать как можно больше о состоянии и доступных методах лечения.
DEBRA — это национальная благотворительная организация, которая предоставляет помощь, советы и поддержку людям в Великобритании, живущим с БЭ.
DEBRA International — всемирная сеть национальных групп, работающих от имени людей, пострадавших от БЭ.
Поддержка лиц, осуществляющих уход
Важно не пренебрегать собственным здоровьем и благополучием при уходе за ребенком со сложным и тяжелым заболеванием, таким как БЭ.
Прочтите о перерывах для лиц, осуществляющих уход, и передышках по уходу и уходу за ребенком-инвалидом.
Информация о вас
Если у вас или вашего ребенка есть БЭ, ваша медицинская бригада передаст информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать лучшие способы профилактики и лечения этого заболевания. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре (GOV. UK)
UK)
Последняя проверка страницы: 23 июня 2021 г.
Дата следующей проверки: 23 июня 2024 г.
Местный буллезный эпидермолиз: MedlinePlus Genetics
Описание
Местный буллезный эпидермолиз (БЭБ) — это основная форма буллезного эпидермолиза, группы генетических состояний, которые делают кожу очень хрупкой и на ней легко образуются волдыри. Волдыри и участки потери кожи (эрозии) образуются в ответ на незначительные травмы или трения, такие как растирание или расчесывание. Исследователи классифицируют соединительный буллезный эпидермолиз на два основных типа: генерализованный тяжелый побёрд (ранее известный как ПБЭ Herlitz) и промежуточный генерализованный ПБЭ (ранее известный как ПБЭ, не относящийся к Herlitz). Хотя типы различаются по степени тяжести, их признаки значительно перекрываются, и они могут быть вызваны мутациями в одних и тех же генах.
Тяжелый генерализованный БЭБ является более серьезной формой заболевания. С рождения или раннего младенчества у больных появляются волдыри на больших участках тела. Волдыри также поражают слизистые оболочки, например, влажную оболочку рта и пищеварительного тракта, что может затруднить прием пищи и переваривание пищи. В результате многие больные дети недоедают и медленно растут. Обширное образование пузырей приводит к рубцеванию и образованию красных неровных пятен, называемых грануляционной тканью. Грануляционная ткань легко и обильно кровоточит, что делает пораженных младенцев восприимчивыми к серьезным инфекциям и потере необходимых белков, минералов и жидкости. Кроме того, скопление грануляционной ткани в дыхательных путях может привести к слабому, хриплому плачу и затрудненному дыханию.
С рождения или раннего младенчества у больных появляются волдыри на больших участках тела. Волдыри также поражают слизистые оболочки, например, влажную оболочку рта и пищеварительного тракта, что может затруднить прием пищи и переваривание пищи. В результате многие больные дети недоедают и медленно растут. Обширное образование пузырей приводит к рубцеванию и образованию красных неровных пятен, называемых грануляционной тканью. Грануляционная ткань легко и обильно кровоточит, что делает пораженных младенцев восприимчивыми к серьезным инфекциям и потере необходимых белков, минералов и жидкости. Кроме того, скопление грануляционной ткани в дыхательных путях может привести к слабому, хриплому плачу и затрудненному дыханию.
Другие осложнения тяжелого генерализованного ПОБЭ могут включать сращение пальцев рук и ног, аномалии ногтей на руках и ногах, деформации суставов (контрактуры), ограничивающие подвижность, выпадение волос (алопеция) и истончение защитного наружного слоя (эмали) зубы.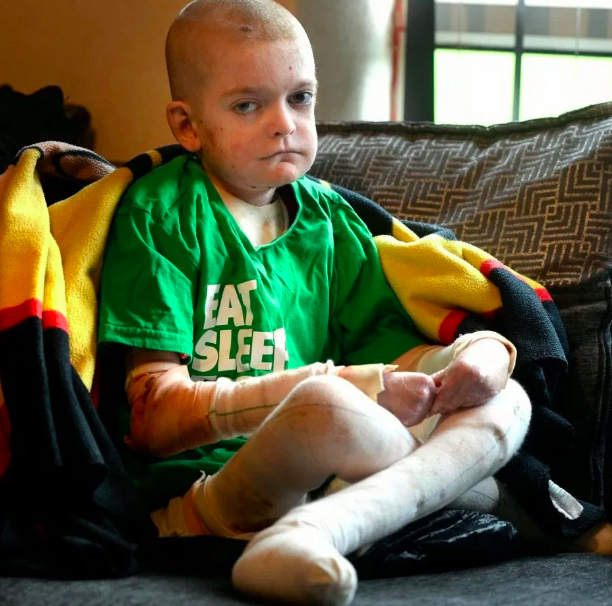 Поскольку признаки и симптомы генерализованного тяжелого БЭБ настолько серьезны, дети с этим заболеванием обычно не выживают после первого года жизни.
Поскольку признаки и симптомы генерализованного тяжелого БЭБ настолько серьезны, дети с этим заболеванием обычно не выживают после первого года жизни.
Более легкая форма пограничного буллезного эпидермолиза называется генерализованным промежуточным пограничным эпидермолизом. Образование пузырей, связанное с генерализованным промежуточным побочным эффектом, может быть ограничено кистями, стопами, коленями и локтями, и оно часто проходит после периода новорожденности. Другие характерные признаки этой формы заболевания включают выпадение волос, аномальные ногти на руках и ногах, а также неровную зубную эмаль. У большинства пораженных людей нет обширных рубцов или образования грануляционной ткани, поэтому затрудненное дыхание и другие тяжелые осложнения встречаются редко. Генерализованный промежуточный вариант JEB обычно ассоциируется с нормальной продолжительностью жизни.
Частота
Оба типа пограничного буллезного эпидермолиза встречаются редко, вместе поражая примерно 3 человека на миллион человек в год в США.
Причины
Пограничный буллезный эпидермолиз чаще всего возникает в результате мутаций в генах LAMA3 , LAMB3 , LAMC2 и COL17A1 . Мутации в каждом из этих генов могут вызывать генерализованный тяжелый ПБЭ или генерализованный промежуточный ПБЭ. Мутации гена LAMB3 являются наиболее распространенными, вызывая около 70 процентов всех случаев буллезного эпидермолиза узлового эпидермолиза.
Гены LAMA3 , LAMB3 и LAMC2 обеспечивают инструкции по созданию одной части (субъединицы) белка, называемого ламинином 332. Этот белок играет важную роль в укреплении и стабилизации кожи, помогая прикреплять верхнего слоя кожи (эпидермиса) к нижележащим слоям. Мутации в любом из трех генов ламинина 332 приводят к продукции дефектной или нефункциональной версии этого белка. Без функционального ламинина 332 клетки эпидермиса хрупкие и легко повреждаются. Трение или другая незначительная травма могут привести к отделению слоев кожи, что приведет к образованию волдырей.
Ген COL17A1 содержит инструкции по созданию белка, используемого для сборки коллагена XVII типа. Коллагены — это молекулы, которые придают структуру и прочность соединительным тканям, таким как кожа, сухожилия и связки, по всему телу. Коллаген XVII типа помогает прикрепить эпидермис к нижележащим слоям кожи, делая кожу прочной и эластичной. Мутации в гене COL17A1 препятствуют нормальному образованию коллагена XVII. В результате кожа менее устойчива к трению и незначительным травмам, а также легко покрывается волдырями. Большинство Мутации гена COL17A1 вызывают генерализованный промежуточный вариант БЭБ, хотя у нескольких лиц с мутациями в этом гене был более серьезный генерализованный тяжелый БЭБ.
Очень редко пограничный буллезный эпидермолиз вызывается мутациями в другом гене, который обеспечивает инструкции по созданию другого белка, помогающего прикреплять верхний слой кожи к нижележащим слоям.
Наследование
Оба типа пограничного буллезного эпидермолиза наследуются по аутосомно-рецессивному типу, что означает, что обе копии гена в каждой клетке имеют мутации.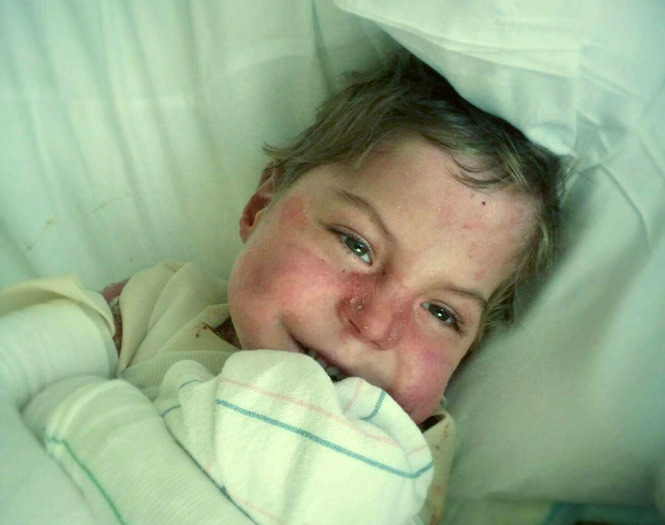 Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания. В редких случаях люди с одной мутировавшей копией 9Гены 0154 COL17A1 , LAMA3 или LAMB3 имеют неровную зубную эмаль.
Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания. В редких случаях люди с одной мутировавшей копией 9Гены 0154 COL17A1 , LAMA3 или LAMB3 имеют неровную зубную эмаль.
Другие названия для этого состояния
- Буллезный эпидермолиз, узловой
- JEB
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: пограничный буллезный эпидермолиз
- Реестр генетического тестирования: пограничный буллезный эпидермолиз Herlitz
- Реестр генетического тестирования: пограничный буллезный эпидермолиз, не относящийся к типу Herlitz
Информационный центр генетических и редких заболеваний
- Буллезный эпидермолиз
- Пограничный буллезный эпидермолиз
Ресурсы поддержки и защиты интересов пациентов
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov
- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- Буллезный эпидермолиз, JUNCTIONAL, HERLITZ TYPE
- ЭПИДЕРМОЛИЗ БУЛЛОЗНЫЙ, СОЕДИНИТЕЛЬНЫЙ, НЕ HERLITZ ТИП
Научные статьи в PubMed
- PubMed
Каталожные номера
- Castori M, Floriddia G, De Luca N, Pascucci M, Ghirri P, Boccaletti V, El
Hachem M, Zambruno G, Castiglia D. Herlitz буллезный эпидермолиз соединительной ткани:
Мутационный профиль ламинина-5 и частота носительства в итальянской популяции. бр
J Дерматол. 2008 г., январь; 158 (1): 38–44. doi: 10.1111/j.1365-2133.2007.08208.x. Epub
2007 г., 4 октября. Цитирование в PubMed 9.0012 - Condrat I, He Y, Cosgarea R, Has C. Пограничный буллезный эпидермолиз: аллельный
Гетерогенность и стратификация мутаций для точной медицины. Фронт Мед
(Лозанна). 2019 29 января; 5:363. doi: 10.3389/fmed.2018.00363. eCollection 2018. Цитирование в PubMed или бесплатная статья в PubMed Central
Цитирование в PubMed или бесплатная статья в PubMed Central - Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, Hintner
Х., Овнанян А., Джонкман М.Ф., Ли И., МакГрат Дж.А., Меллерио Дж.Е., Мюррелл Д.Ф., Симидзу
Х., Уитто Дж., Валквист А., Вудли Д., Замбруно Г. Классификация унаследованных
буллезный эпидермолиз (БЭ): отчет Третьего международного консенсусного совещания
по диагностике и классификации БЭ. J Am Acad Дерматол. 2008 июнь; 58 (6): 931-50.
doi: 10.1016/j.jaad.2008.02.004. Epub 2008, 18 апреля. Цитирование на PubMed - Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escamez MJ, Fuentes I,
Бухель С., Хиремагалор Р., Пола-Губо Г., ван ден Аккер П.С., Вертхайм-Тисаровска К.,
Замбруно Г. Клинические рекомендации по лабораторной диагностике
буллезный эпидермолиз. Бр Дж Дерматол. 2020 март; 182(3):574-592. дои:
10.1111/bjd.18128. Epub 2019, 9 августа. Цитирование в PubMed или бесплатная статья на PubMed Central - Келли-Манкузо Г.
 , Копелан Б., Азизхан Р.Г., Лаки А.В. Узловой эпидермолиз
, Копелан Б., Азизхан Р.Г., Лаки А.В. Узловой эпидермолиз
Bullosa заболеваемость и выживаемость: 5-летний опыт дистрофического эпидермолиза
Преподаватель медсестер Американской исследовательской ассоциации Bullosa (DebRA), с 2007 по 2011 год.
Педиатр Дерматол. 2014 март-апрель;31(2):159-62. doi: 10.1111/pde.12157. Электронная книга 2013 г.
31 мая. Цитирование в PubMed - Muhle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L, Meneguzzi G, Schneider
H. Новые и повторяющиеся мутации в генах ламинина-5, вызывающие летальный исход
Буллезный эпидермолиз: молекулярные основы и клиническое течение болезни Герлица.
Хам Жене. 2005 г., январь; 116 (1–2): 33–42. дои: 10.1007/s00439-004-1210-й. Epub, ноябрь 2004 г.
5. Цитирование в PubMed - Накано А., Лестрингант Г.Г., Паперна Т., Бергман Р., Гершони Р., Фроссар П., Канаан
М., Менегуцци Г., Ричард Г., Пфенднер Э., Уитто Дж., Пулккинен Л., Спречер Э.
Пограничный буллезный эпидермолиз на Ближнем Востоке: клинические и генетические исследования
в ряду кровнородственных семей.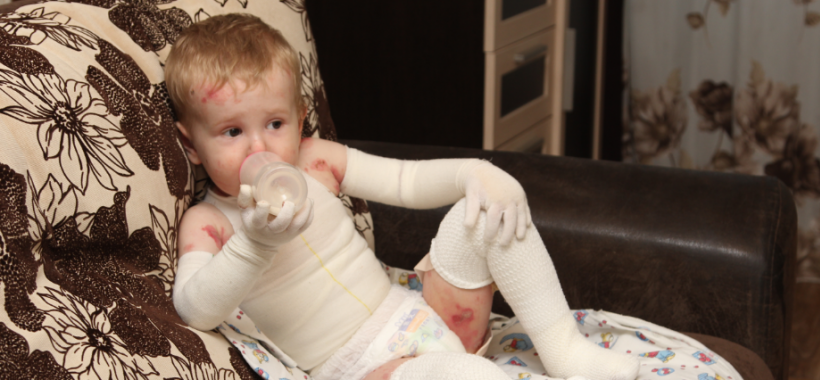 J Am Acad Дерматол. 2002 г., апрель; 46 (4): 510-6.
J Am Acad Дерматол. 2002 г., апрель; 46 (4): 510-6.
doi: 10.1067/mjd.2002.119673. Цитата в PubMed - Пфенднер Э.Г., Брукнер А., Конгет П., Меллерио Дж., Палиссон Ф., Лаки А.В. Базовый
наука о буллезном эпидермолизе, диагностика и молекулярная характеристика:
Материалы II Международного симпозиума по буллезному эпидермолизу,
Сантьяго, Чили, 2005 г. Int J Dermatol. 2007 авг; 46 (8): 781-94. Дои:
10.1111/j.1365-4632.2007.03307.х. Аннотация недоступна. Цитата в PubMed - Пфенднер Э.Г., Лаки А.В. Пограничный буллезный эпидермолиз. 2008 фев 22 [обновлено
2018 20 декабря]. Пришли: Адам MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH,
Грипп К.В., Амемия А., редакторы. GeneReviews(R) [Интернет]. Сиэтл (Вашингтон):
Вашингтонский университет, Сиэтл; 1993-2023 гг. Доступна с
http://www.ncbi.nlm.nih.gov/books/NBK1125/
Цитата в PubMed - Пулккинен Л., Уитто Дж. Анализ мутаций и молекулярная генетика
буллезный эпидермолиз.
.