
Спинальная мышечная атрофия симптомы у детей: что это такое? — Про Паллиатив

Спинальная мышечная атрофия
Наука
Спинальная мышечная атрофия (СМА) — генетическое заболевание, характеризующееся развитием прогрессирующей мышечной слабости.
СМА возникает из-за мутации гена SMN1, которая приводит к нарушению работы двигательных нейронов спинного мозга — особых клеток, которые отвечают за передачу сигнала от нервной системы к мышцам. В результате развивается нарастающая мышечная слабость конечностей. Из-за того, что мышцы теряют связь с нервными клетками, происходит гипотрофия мышц (то есть теряется мышечная масса). Кроме того, заболевание также характеризуется скелетными нарушениями (сколиозом, деформациями грудной клетки), из-за ограничения подвижности суставов развиваются контрактуры, постепенно развивается дыхательная недостаточность. Интеллект при СМА остается полностью сохранным и развивается так же, как у здоровых людей.
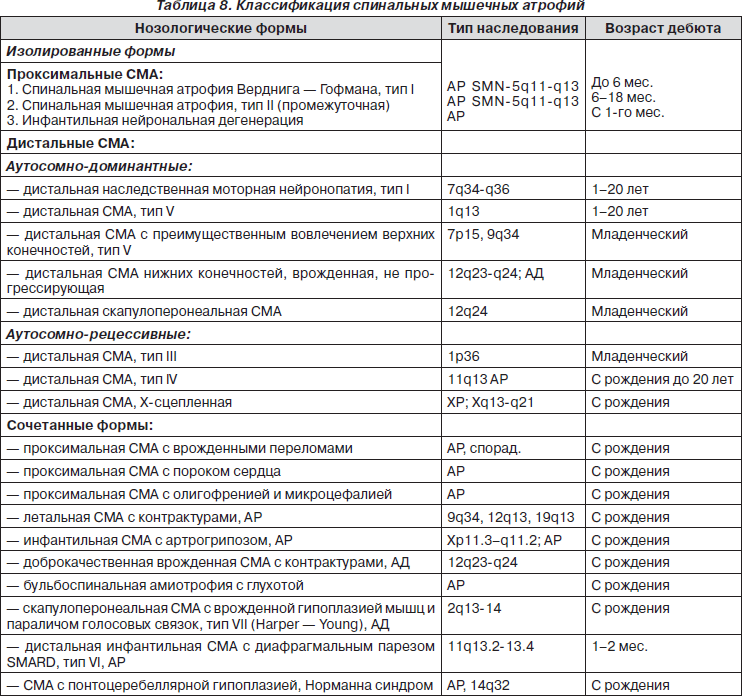
Выделяют несколько основных типов СМА, которые отличаются по выраженности проявлений и возрасту начала заболевания. СМА 1 типа (болезнь Верднига-Гоффмана) начинается в возрасте <6 месяцев и характеризуется наиболее тяжелым течением заболевания. СМА 2 типа (болезнь Дубовица) начинается позже, в возрасте 7-18 месяцев и характеризуется более медленным течением. СМА 3 типа (болезнь Кугельберга-Веландера) характеризуется началом после 18 месяцев. Дети учатся самостоятельно сидеть и ходить, при этом наблюдаются проблемы с более сложными двигательными актами — бегом, подъёмом по лестнице и др. Мышечная слабость в большинстве случаев прогрессирует медленно. Часть пациентов доживает до взрослого возраста, сохраняя способность к передвижению, другие к подростковому возрасту нуждаются в инвалидной коляске. Со временем могут возникнуть проблемы с глотанием, откашливанием, дыханием. При надлежащем уходе пациенты имеют обычную продолжительность жизни. Крайне редко развивается СМА 4 типа, когда заболевание манифестирует уже во взрослом возрасте (обычно на 2-3 десятилетии жизни). Нарастание слабости очень медленное, продолжительность жизни не изменяется.
СМА 1 типа (болезнь Верднига-Гоффмана) начинается в возрасте <6 месяцев и характеризуется наиболее тяжелым течением заболевания. СМА 2 типа (болезнь Дубовица) начинается позже, в возрасте 7-18 месяцев и характеризуется более медленным течением. СМА 3 типа (болезнь Кугельберга-Веландера) характеризуется началом после 18 месяцев. Дети учатся самостоятельно сидеть и ходить, при этом наблюдаются проблемы с более сложными двигательными актами — бегом, подъёмом по лестнице и др. Мышечная слабость в большинстве случаев прогрессирует медленно. Часть пациентов доживает до взрослого возраста, сохраняя способность к передвижению, другие к подростковому возрасту нуждаются в инвалидной коляске. Со временем могут возникнуть проблемы с глотанием, откашливанием, дыханием. При надлежащем уходе пациенты имеют обычную продолжительность жизни. Крайне редко развивается СМА 4 типа, когда заболевание манифестирует уже во взрослом возрасте (обычно на 2-3 десятилетии жизни). Нарастание слабости очень медленное, продолжительность жизни не изменяется.
Почему у разных людей возникают разные типы СМА?
На настоящий момент известно, что тяжесть проявлений зависит от количества копий гена SMN2. Этот ген частично замещает выработку белка SMN, которого недостает при СМА. Поэтому чем больше копий этого гена, тем позже дебют заболевания и тем медленнее развиваются симптомы. Скорее всего, существуют и другие механизмы, о которых мы пока еще не знаем, в мире продолжается работа по их изучению.
На основании чего ставится диагноз СМА?
Для подтверждения диагноза необходимо проведение генетического анализа на определение мутации в гене SMN1. Кроме того, проводится анализ на определение количества копий гена SMN2 — это может помочь прогнозировать тяжесть заболевания и определить эффективность некоторых лекарственных средств. Оба анализа можно сдать на базе нашего Центра (см. раздел ДНК-диагностика).
Кроме этого, может понадобиться проведение игольчатой электромиографии, которая выявляет поражение двигательных нейронов и поможет исключить другие нервно-мышечные заболевания.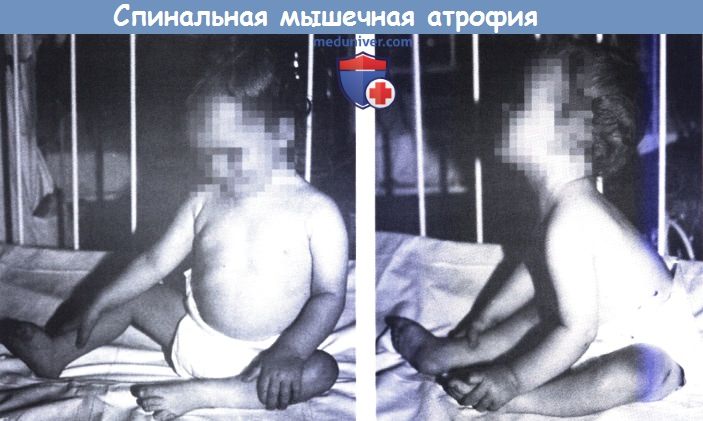 Однако данные этого исследования неспецифичны, так как поражение двигательных нейронов встречается и при других заболеваниях.
Однако данные этого исследования неспецифичны, так как поражение двигательных нейронов встречается и при других заболеваниях.
Какие есть возможности лечения?
Помимо поддерживающий и симптоматической терапии в последнее время появились препараты, специально разработанные для лечения данного заболевания, и которые могут увеличивать синтез необходимого белка. Наши специалисты помогут подобрать необходимую терапию с учетом всех показаний и противопоказаний.
Кроме специфической терапии большое значение у пациентов со СМА имеет правильный уход и реабилитация.
5 неврологическое отделение ФГБНУ «Научный центр неврологии»
16.07.2021
Поделиться ссылкой:
что это такое, виды и генетические особенности заболевания
Опухоль шеи: симптомы, диагностика, лечение
12 мая, 2023
Генетические анализы при планировании беременности
28 апреля, 2023
Безлактозная диета
9 апреля, 2023
Синдром Мартина-Белл
29 марта, 2023
Витамин Е
27 марта, 2023
Тест ДНК во время беременности
26 марта, 2023
Смотреть все
Исследование рецептивности
эндометрия
14 августа 2017 г
Содержание:
- Виды заболевания
- Причины
- Диагностика СМА
- Лечение спинальной мышечной атрофии
Одним из наиболее частых аутосомно-рецессивных заболеваний у детей является спинальная мышечная атрофия (СМА).
Спинальная мышечная атрофия – наследственное нервно-мышечное заболевание, причиной которого является постепенное и необратимое нарушение функций двигательных нейронов спинного мозга, приводящее к симметричному ослабеванию, а затем атрофии мышц. Хотя это заболевание относится к редким (орфанным), оно является вторым (после муковисцидоза) по распространенности из наследственных среди коренных европейцев и населения Северной Америки европейского происхождения.
Виды заболевания
Различают четыре типа СМА, в зависимости от возраста манифестации и степени проявления симптомов:
- СМА 1 типа (младенческая форма), или болезнь Верднига-Гоффмана – самая ранняя и тяжелая форма заболевания. Обычный возраст начала 0-6 месяцев. Чаще всего дети с такой формой заболевания не доживают до 2-х лет.
- СМА 2 типа (промежуточная форма), или болезнь Дубовица – возраст проявления этой формы заболевания 6-18 месяцев. Дети с таким типом СМА могут сидеть без поддержки, но не могут вставать и ходить.
 Тяжесть заболевания и прогноз зависит от скорости вовлечения в болезнь мышц, отвечающих за дыхание.
Тяжесть заболевания и прогноз зависит от скорости вовлечения в болезнь мышц, отвечающих за дыхание. - СМА 3 типа (ювенильная форма), болезнь Кугельберга-Веландер – проявляется в возрасте старше 18 месяцев. Дети с этой формой СМА способны сравнительно длительное время ходить самостоятельно, слабость и атрофия мышц прогрессируют медленно.
- СМА 4 типа (взрослая форма) — самая легкая форма заболевания, обычно проявляющаяся на втором или третьем десятилетии жизни симптомами, аналогичными ювенильной форме. Как правило, не приводит к снижению продолжительности жизни пациента.
Причины
Генетической причиной СМА являются мутации в гене SMN1. Он кодирует белок, необходимый для работы двигательных нейронов. Важной частью этого белка является фрагмент, информация о котором содержится в 7 экзоне (определенной части) гена SMN1. Любая мутация, которая приводит к отсутствию этого фрагмента в белке или же значительно нарушает строение белка, может привести к развитию заболевания. Полное отсутствие 7 экзона (делеция) является самой частой причиной СМА. Если на обеих хромосомах в гене SMN1 есть нарушения в 7 экзоне, то нормальный белок не будет формироваться, а отсутствие нормально функционирующего белка вызывает СМА.
Полное отсутствие 7 экзона (делеция) является самой частой причиной СМА. Если на обеих хромосомах в гене SMN1 есть нарушения в 7 экзоне, то нормальный белок не будет формироваться, а отсутствие нормально функционирующего белка вызывает СМА.
В геноме человека также есть ген SMN2. Он очень похож на ген SMN1 (99% совпадения последовательностей). Одно из ключевых отличий этих двух генов – определенный нуклеотид в 7 экзоне. Одна замена в последовательности гена SMN2 приводит к тому, что с него синтезируется нефункциональный белок. Однако, в результате ошибок синтеза белка, которые иногда случаются в нашем организме, с гена SMN2 может синтезироваться небольшое количество правильного, функционального белка, необходимого для работы двигательных нейронов. Именно поэтому даже при полном отсутствии нормального белка с гена SMN1 количество копий гена SMN2 влияет на тяжесть симптомов, однако мутации SMN2 сами по себе не могут быть причиной СМА и не имеют клинического значения, если не нарушена функция SMN1.
Диагностика СМА
Информация о количестве копий генов SMN1 и SMN2 позволяет диагностировать заболевание и оценить риск рождения в семье ребенка с диагнозом СМА. Если оба родителя являются носителями мутации в гене SMN1, вероятность рождения больного ребенка равна 25%.
Молекулярная диагностика носительства СМА и самого заболевания осложнена схожестью генов SMN1 и SMN2. Для диагностики необходимо использовать методы, позволяющие различить эти гены по немногим отличающимся точкам. Так как ключевую роль играет именно экзон 7, необходимо использовать метод, который позволяет определить количество его копий в гене SMN1.
Сегодня одним из наиболее надежных методов точного определения количества копий каждого гена является анализ методом MLPA (мультиплексная амплификация лигазно-связанных проб). В результате анализа лаборатория получает заключение о количестве 7 и 8 экзонов генов SMN1 и SMN2. Хотя у большинства пациентов со СМА выявляются делеции 7 и 8 экзона в гене SMN1, для молекулярного подтверждения диагноза ориентируются только на данные по количеству копий 7 экзона.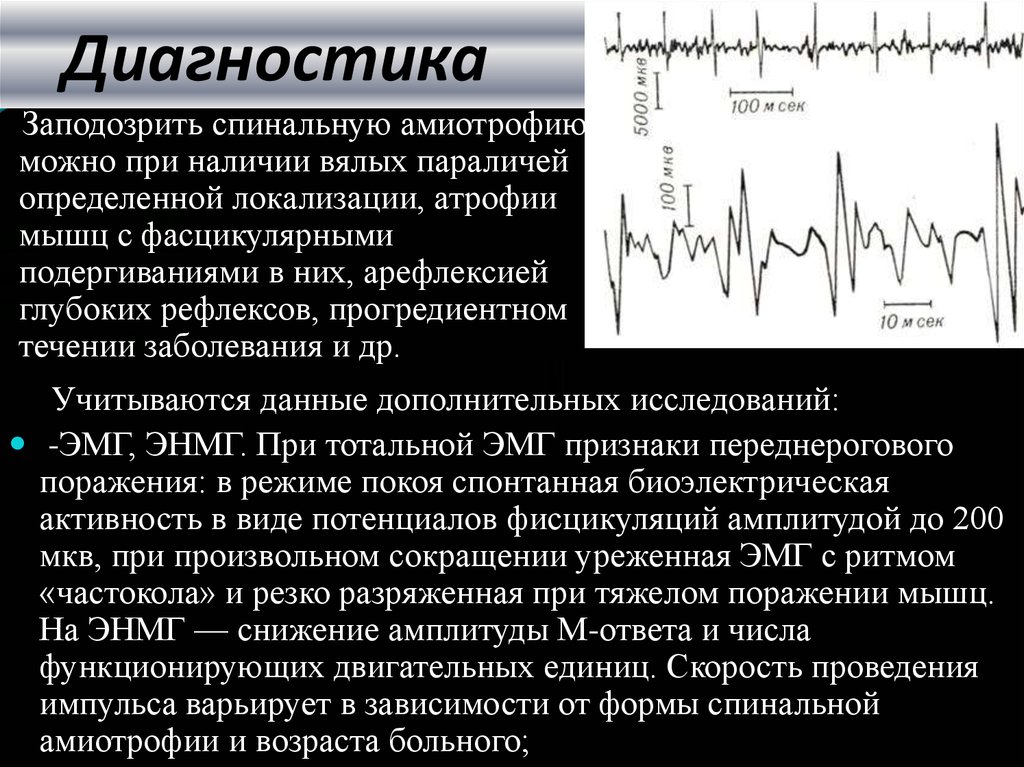
Спинальная амиотрофия типы I, II, III, IV: поиск делеций в гене SMN1
В России каждый 35-36-й человек является бессимптомным носителем мутации в гене SMN1.
Перейти к услуге
Лечение спинальной мышечной атрофии
В настоящее время в мире существуют 3 препарата, позволяющих замедлить прогрессирование СМА, все они зарегистрированы в РФ, являются дорогостоящими: «Спинраза» (Biogen) – первое лекарство, которое было одобрено для лечения СМА, требуется курс инъекций, препарат включен в российские клинические рекомендации по лечению СМА и перечень ЖНВЛП; «Золгенсма» (Novartis) – достаточно 1 инъекции, препарат некоторое время считался самым дорогим лекарством в мире; «Эврисди» (Roche) – препарат для перорального применения. Ряд других препаратов находится на стадии клинических исследований, в том числе препараты российских производителей (например, ANB-4 от Biocad).
Поскольку на данный момент неизвестно, можно ли полностью вылечить СМА, и существующие лекарства нацелены в первую очередь на замедление и возможную остановку прогрессирования заболевания, многим семьям важно иметь возможность снизить риск рождения больного ребенка в семье. Для этого необходимо проводить генотипирование будущих родителей, которое позволит оценить этот риск. В случае выявления статуса носительства мутаций в гене SMN1 обоими родителями требуется консультация врача-генетика. Врач может предложить различные способы снижения риска для потомства, в частности, паре может быть рекомендована пренатальная или преимплантационная генетическая диагностика СМА.
Для этого необходимо проводить генотипирование будущих родителей, которое позволит оценить этот риск. В случае выявления статуса носительства мутаций в гене SMN1 обоими родителями требуется консультация врача-генетика. Врач может предложить различные способы снижения риска для потомства, в частности, паре может быть рекомендована пренатальная или преимплантационная генетическая диагностика СМА.
Список используемой литературы:
- Prior, T., et al. Technical standards and guidelines for spinal muscular atrophy testing. Genet Med 13, 686–694 (2011). https://doi.org/10.1097/GIM.0b013e318220d523.
- Prior TW, et al. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2020 Dec 3]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352/
- Gregg, A.R., et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG).
 Genet Med 23, 1793–1806 (2021). https://doi.org/10.1038/s41436-021-01203-z
Genet Med 23, 1793–1806 (2021). https://doi.org/10.1038/s41436-021-01203-z - Burghes A. H. M., Beattie C.E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? // Nat Rev Neurosci. 2009 V. 10(8) P. 597-609.
- Su Y. N. et al. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009: a prospective population-based cohort study //PloS one.2011;6(2):e17067.
- Sugarman E. A. et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of> 72 400 specimens //European journal of human genetics.2012;20(1):27.
- Tisdale S., Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy //Journal of Neuroscience.2015;35(23):8691-8700.
- Забненкова В. В., Дадали Е. Л., Поляков А. В. Проксимальная спинальная мышечная атрофия типов I–IV: особенности молекулярно-генетической диагностики //Нервно-мышечные болезни – 2013.
 – № 3. – С.27-31.
– № 3. – С.27-31. - Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. и др. Молекулярно-генетический анализ наследственных нейро дегенеративных заболеваний // Генетика. 2004. Т. 40. № 6.
- Клюшников С.А., Иллариошкин С.Н., Иванова-Смоленская И.А. Семейный случай спинально-бульбарной амиотрофии Кеннеди // Нервные болезни. 2008. № 1.
Спинальная мышечная атрофия (СМА) | Бостонская детская больница
Как диагностируется спинальная мышечная атрофия?
СМА иногда трудно диагностировать, поскольку симптомы могут напоминать другие состояния или проблемы со здоровьем. Врачи обычно диагностируют СМА после того, как у ребенка появляется мышечная слабость и сниженный мышечный тонус.
Если ваш врач подозревает СМА, он может использовать следующие тесты для диагностики состояния:
- генетические анализы крови, которые могут подтвердить диагноз СМА
- электромиографический (ЭМГ) тест, который измеряет электрическую активность мышцы или группы мышц (в некоторых случаях)
- тест на креатинкиназу (КФК) (при необходимости отличить от других типов нервно-мышечных заболеваний)
Какие существуют варианты лечения спинальной мышечной атрофии?
Пока полного излечения от СМА не существует. Однако открытие генетической причины СМА привело к разработке нескольких вариантов лечения, которые воздействуют на гены, участвующие в СМА, — заместительная терапия генами под названием Золгенсма и два препарата, называемые нусинерсен (Спинраза) и рисдиплам (Эвирсди). Все три метода лечения прошли клинические испытания в Бостонской детской больнице и в других местах, прежде чем они были одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
Однако открытие генетической причины СМА привело к разработке нескольких вариантов лечения, которые воздействуют на гены, участвующие в СМА, — заместительная терапия генами под названием Золгенсма и два препарата, называемые нусинерсен (Спинраза) и рисдиплам (Эвирсди). Все три метода лечения прошли клинические испытания в Бостонской детской больнице и в других местах, прежде чем они были одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
Вы и команда по уходу за вашим ребенком вместе примете решение о наиболее подходящем плане лечения на основе нескольких факторов:
- возраст вашего ребенка, состояние здоровья и медицинские потребности
- SMA тип
- степень и тип симптомов
- способность вашего ребенка переносить процедуры и лечение
- семейные предпочтения
Генная терапия СМА (Zolgensma®)
Boston Children’s — одна из первых детских больниц в стране, которая предлагает одобренную FDA генную терапию для лечения спинальной мышечной атрофии у детей младше 2 лет. Эта генная терапия, известная под торговой маркой Zolgensma®, предотвращает дальнейшую дегенерацию двигательных нейронов и мышц путем замены дефектных или отсутствующих 9 нейронов.0037 SMN1 ген.
Эта генная терапия, известная под торговой маркой Zolgensma®, предотвращает дальнейшую дегенерацию двигательных нейронов и мышц путем замены дефектных или отсутствующих 9 нейронов.0037 SMN1 ген.
Известный ранее как AVXS-101, этот одноразовый метод генной терапии заменяет дефектный или отсутствующий ген ( SMN1 ), который отвечает за выработку белка двигательных нейронов выживания (SMN). Предоставление функциональной копии SMN1 предотвращает дальнейшее вырождение двигательных нейронов и мышц.
Перед генной терапией дети сначала сдают анализы крови, чтобы убедиться, что пациенты подходят для лечения. Zolgensma® вводится внутривенно (в/в) в нашем лечебном центре. Инфузия занимает около часа.
Для получения дополнительной информации о генной терапии СМА или для направления пациента обращайтесь в Программу спинальной мышечной атрофии по телефону 617-919-6814.
Рисдиплам (Evrysdi)
С 2016 года Бостонская детская программа по борьбе со спинальной мышечной атрофией активно участвует в ключевых клинических испытаниях рисдиплама (торговая марка Evrysdi), первого перорального препарата для лечения СМА. Рисдиплам является третьим препаратом для лечения СМА и был одобрен FDA в 2020 году для использования у детей от 2 месяцев и старше.
Рисдиплам является третьим препаратом для лечения СМА и был одобрен FDA в 2020 году для использования у детей от 2 месяцев и старше.
Наша команда входит в число исследователей, участвующих в продолжающемся испытании FIREFISH, которое предоставило некоторую клиническую информацию, которая привела к одобрению ридсиплама FDA. Это и другие испытания, в которых мы участвуем, продолжат исследовать рисдиплам у участников в течение нескольких лет.
Nusinersen (Spinraza)
Наша программа также участвовала в клинических испытаниях nusinersen (торговая марка Spinraza), первого препарата для лечения СМА. Мы были первой больницей в мире, включившей младенца со СМА 1 типа в исследование ENDEAR фазы 3 в 2014 году. , и мы начали предлагать препарат всем подходящим пациентам со СМА. Наша многопрофильная команда специалистов по СМА помогла разработать стандартизированные способы измерения симптомов СМА у пациентов и изменения этих симптомов с течением времени. Эти результаты используются для измерения эффективности нусинерсена и других методов лечения.
Как мы лечим спинальную мышечную атрофию
Программа лечения спинальной мышечной атрофии в Бостонской детской больнице объединяет группу экспертов разных специальностей, имеющих опыт ухода за детьми со СМА. Один или два раза в месяц мы организуем специальную клинику со СМА, чтобы ваш ребенок мог получить всю необходимую помощь, связанную со СМА, за одно посещение. Такой подход означает, что наши различные специалисты могут тесно сотрудничать с вашей семьей, чтобы обеспечить хорошую координацию ухода за вашим ребенком. Наша программа предлагает несколько вариантов лечения СМА, включая генную терапию.
Спинальная мышечная атрофия
Спинальная мышечная атрофия (СМА) представляет собой группу генетических (передаваемых родителями) заболеваний, которые поражают двигательные нейроны (нервные клетки) в спинном мозге, вызывая ослабление произвольных мышц (мышц, которыми вы управляете) . Это может повлиять на ползание, ходьбу, глотание, дыхание и другие функции. Интеллектуальное развитие при СМА является нормальным. На самом деле люди со СМА часто являются высокоинтеллектуальными людьми.
Интеллектуальное развитие при СМА является нормальным. На самом деле люди со СМА часто являются высокоинтеллектуальными людьми.
Основные формы спинальной мышечной атрофии
Существует три основных типа СМА, поражающих младенцев и детей:
- СМА I типа (болезнь Верднига-Гоффмана)
- SMA тип II
- СМА III типа (болезнь Кугельберга-Веландера)
СМА I типа
СМА I типа, также известная как болезнь Верднига-Гоффмана, является наиболее тяжелой формой СМА. Это происходит между рождением и шестимесячным возрастом. Для этих больных характерно «колокольное» телосложение с узкой впалой грудной клеткой и большим животом. Десять процентов новорожденных с тяжелым типом СМА I рождаются с тугоподвижными суставами (врожденными контрактурами). Их руки могут оставаться сжатыми в кулаки, а ноги могут быть зафиксированы в положении «лягушки». Пораженные младенцы сильно гипотоничны (вялые) с чрезвычайно слабыми мышцами всего тела. Они очень мало двигаются, не контролируют голову, у них могут быть серьезные проблемы с дыханием и неспособность есть. Он также поражает язык, лицо и мышцы челюсти, а также мышцы пищеварительного тракта. Поэтому запоры, плохое питание и слюнотечение являются обычным явлением. Слабость дыхательных мышц часто приводит к пневмонии, которая может быть опасной для жизни. Дыхательные мышцы могут стать настолько слабыми, что пациенты не смогут дышать самостоятельно, и может быть использована искусственная вентиляция легких (респираторная поддержка жизни). Ожидаемая продолжительность жизни детей со СМА I типа сокращается из-за множества сопутствующих осложнений заболевания. Согласно текущей статистике, более двух третей этих детей умирают в возрасте до двух лет.
Они очень мало двигаются, не контролируют голову, у них могут быть серьезные проблемы с дыханием и неспособность есть. Он также поражает язык, лицо и мышцы челюсти, а также мышцы пищеварительного тракта. Поэтому запоры, плохое питание и слюнотечение являются обычным явлением. Слабость дыхательных мышц часто приводит к пневмонии, которая может быть опасной для жизни. Дыхательные мышцы могут стать настолько слабыми, что пациенты не смогут дышать самостоятельно, и может быть использована искусственная вентиляция легких (респираторная поддержка жизни). Ожидаемая продолжительность жизни детей со СМА I типа сокращается из-за множества сопутствующих осложнений заболевания. Согласно текущей статистике, более двух третей этих детей умирают в возрасте до двух лет.
СМА II типа
СМА II типа обычно проявляется в позднем младенчестве, обычно в возрасте от семи до восемнадцати месяцев, и прогрессирует (ухудшается) медленнее, чем СМА I типа. Младенцы со СМА II типа обычно способны сосать и глотать, и не имеют серьезных проблем с дыханием на ранней стадии. Эти дети могут сидеть самостоятельно. Они также могут развить способность стоять при поддержке другого человека, скоб или специального устройства для стояния. Люди с этой формой СМА будут иметь прогрессирующую (ухудшающуюся) слабость и со временем могут оказаться прикованными к инвалидному креслу. Поскольку мышцы между ребрами (межреберные мышцы) ослабевают, у пациентов возникают трудности с кашлем и глубоким вдохом, а также повышается риск респираторных инфекций. Прочность костей снижается, поскольку пациенты не могут стоять, и они подвержены риску переломов костей (переломов). Кроме того, по мере взросления этих детей почти всегда развивается сколиоз (искривление позвоночника). Продолжительность жизни может выходить за рамки школьных лет.
Эти дети могут сидеть самостоятельно. Они также могут развить способность стоять при поддержке другого человека, скоб или специального устройства для стояния. Люди с этой формой СМА будут иметь прогрессирующую (ухудшающуюся) слабость и со временем могут оказаться прикованными к инвалидному креслу. Поскольку мышцы между ребрами (межреберные мышцы) ослабевают, у пациентов возникают трудности с кашлем и глубоким вдохом, а также повышается риск респираторных инфекций. Прочность костей снижается, поскольку пациенты не могут стоять, и они подвержены риску переломов костей (переломов). Кроме того, по мере взросления этих детей почти всегда развивается сколиоз (искривление позвоночника). Продолжительность жизни может выходить за рамки школьных лет.
СМА типа III
Также известная как болезнь Кугельберга-Веландера, это самая легкая форма СМА. В младенчестве могут отсутствовать признаки или симптомы, и пациенты обычно достигают вех раннего развития в соответствующее время. Признаки СМА типа III могут впервые появиться где-то в возрасте от одного года до подросткового возраста.