
Синдром ушера: Синдром Ушера. Что такое Синдром Ушера?

Синдром Ашера (Ушера) | Межрегиональная общественная организация Чтобы видеть!
Справочник
28.10.2016
312
Синдром Ашера (Ушера) — это наследственная болезнь, характеризующаяся ухудшение слуха и прогрессирующим снижением зрения. Изменения зрения происходят из-за пигментного ретинита — дегенеративного заболевания сетчатки глаза, которое обычно проявляется в юности или на ранней стадии взросления. Иногда заболевание оказывает влияние и на вестибулярные функции человека. Проявления болезни могут меняться индивидуальным образом, а его прогресс может идти с различной скоростью.
Существует как минимум три различных формы синдрома Ашера. Первый тип связан с врожденной значительной тугоухостью и нарушением вестибулярных функций. Первые признаки пигментного ретинита — ночная слепота и ухудшение периферийного зрения — обычно наступают еще в юном возрасте.
При синдроме Ашера второго типа новорожденные дети отличаются средним или существенным нарушением слуха. Симптомы пигментного ретинита обычно начинают проявляться вскоре после периода отрочества. Ухудшение зрения происходит не так быстро, как в случае синдрома Ашера первого типа, а тугоухость обычно остается на стабильном уровне и не прогрессирует.
Симптомы пигментного ретинита обычно начинают проявляться вскоре после периода отрочества. Ухудшение зрения происходит не так быстро, как в случае синдрома Ашера первого типа, а тугоухость обычно остается на стабильном уровне и не прогрессирует.
Более редкий синдром Ашера третьего типа был впервые задокументирован в 1995 году. Дети с этим заболеванием обычно рождаются с нормальным слухом или лишь с небольшим его нарушением. Прогрессирующее ухудшение слуха и зрения начинается лишь в возрасте полового созревания. Во многих случаях начинаются и вестибулярные нарушения.
Нарушения слуха при синдроме Ашера возникают из-за генетической мутации, оказывающей влияние на нервные клетки в ушной улитке, которая отвечает за слуховую часть внутреннего уха, воспринимающего и распознающего звуки. Тот же самый генетический дефект негативно влияет и на клетки фоторецепторов в сетчатке, что приводит к ухудшению зрения. Сетчатка — это тонкий слой ткани в задней части глаза, содержащий воспринимающие свет фоторецепторные клетки. Эти клетки, их еще иногда называют колбочки и палочки, отвечают за преобразование света в электрические импульсы, которые передают информацию о том, что находится в поле зрения, в мозг человека.
Эти клетки, их еще иногда называют колбочки и палочки, отвечают за преобразование света в электрические импульсы, которые передают информацию о том, что находится в поле зрения, в мозг человека.
Как наследуется синдром Ашера?
Синдром Ашера передается от родителей к их детям как аутосомно-рецессивная характеристика. При таком типе наследования каждый из родителей является носителем мутировавшего гена, но у них самих это никак не проявляется, поскольку каждый из них несет в себе только одну копию поврежденного гена. Ребенок имеет 25%-вероятность унаследовать мутировавший ген и заболеть. То есть у ребёнка проявится наследственный признак, если он унаследует две повреждённые (мутировавшие) копии гена: одну копию от матери, вторую от отца. Для того, чтобы понять, как происходит наследование этого заболевания, можно проконсультироваться у специалиста по генетике, обсудить с ним такие темы, как планирование семьи, генетическое тестирование и т.п.
Во всем мире синдром Ашера является одной из основных причин возникновения комбинированной слепоты и глухоты. Примерно 30% людей с пигментным ретинитом отмечают у себя частичную потерю слуха, и примерно половина из них имеет синдром Ашера. Чтобы оценить свое состояние и риск для других членов семьи или будущих детей заболеть синдромом Ашера, можно пройти генетическое тестирование, которое в нашей стране уже стало вполне доступной процедурой.
Примерно 30% людей с пигментным ретинитом отмечают у себя частичную потерю слуха, и примерно половина из них имеет синдром Ашера. Чтобы оценить свое состояние и риск для других членов семьи или будущих детей заболеть синдромом Ашера, можно пройти генетическое тестирование, которое в нашей стране уже стало вполне доступной процедурой.
Методы лечения синдрома Ашера
Хотя сегодня и не существует лечения этого заболевания, в настоящее время ведутся интенсивные исследования, которые ставят своей целью нахождение лечения для всех типов синдрома Ашера.
Ученые уже обнаружили ряд генетических изменений, вызывающих эту болезнь и отвечающих за определение различных его подтипов (т.е.1A, 1B, 1C, 1D, 1E, 1F, 1G, 2A, 2B, 2C и 3A). Сегодня ученые исследуют возможность разработки терапии с помощью замены дефектных генов, связанных с синдромом Ашера, на здоровые.
Исследователи также определили вид диеты, которая предположительно замедляет скорость потери зрения у некоторых пациентов с пигментным ретинитом. Хотя это, конечно, не лечение, но это важное открытие – похоже, что ретинол пальмитат (витамин А) может замедлять дегенерацию сетчатки у некоторых людей с пигментным ретинитом и синдромом Ашера 2 типа. Кроме того, при исследованиях выяснилось, что в некоторых случаях докозагексаеновая кислота (ДГК) — полиненасыщенная жирная кислота класса Омега-3 — может усиливать эффект от приема витамина А. Однако, такой эффект от приема ДГК отмечается только в течение первых двух лет терапии с помощью ретинола пальмитата (отметим, что исследования проводились только со взрослыми пациентами, страдающими пигментным ретинитом и синдромом Ашера второго типа, поэтому влияние на других пациентов с синдромом Ашера неизвестно).
Хотя это, конечно, не лечение, но это важное открытие – похоже, что ретинол пальмитат (витамин А) может замедлять дегенерацию сетчатки у некоторых людей с пигментным ретинитом и синдромом Ашера 2 типа. Кроме того, при исследованиях выяснилось, что в некоторых случаях докозагексаеновая кислота (ДГК) — полиненасыщенная жирная кислота класса Омега-3 — может усиливать эффект от приема витамина А. Однако, такой эффект от приема ДГК отмечается только в течение первых двух лет терапии с помощью ретинола пальмитата (отметим, что исследования проводились только со взрослыми пациентами, страдающими пигментным ретинитом и синдромом Ашера второго типа, поэтому влияние на других пациентов с синдромом Ашера неизвестно).
Кроме исследования диетической терапии, ученые работают и над другими вариантами потенциального лечения этого заболевания, включая использование искусственных имплантатов сетчатки, генную терапию и стволовые клетки.
Сопутствующие заболевания
Существует ряд заболеваний, некоторые из которых являются также наследственными, которые являются следствием глухоты или комбинации глухоты и слепоты, но они не имеют отношения к синдрому Ашера. Однако, пигментный ретинит, связанный с синдромом Ашера, имеет характеристики, большинство из которых такие же, как у других форм пигментного ретинита. И ученые надеются, что успехи, достигнутые в понимании природы и лечении других форм пигментного ретинита, принесут непосредственную пользу и людям с синдромом Ашера, и наоборот.
Однако, пигментный ретинит, связанный с синдромом Ашера, имеет характеристики, большинство из которых такие же, как у других форм пигментного ретинита. И ученые надеются, что успехи, достигнутые в понимании природы и лечении других форм пигментного ретинита, принесут непосредственную пользу и людям с синдромом Ашера, и наоборот.
Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов | Иванова М.Е., Атарщиков Д.С., Демчинский А.М., Стрельников В.В., Бар Д., Порядин Г.В., Балашова Л.М., Салмаси Ж.М.
В статье представлены результаты исследования, посвященного изучению спектра мутаций российской когорты пациентов с синдромом Ашера и проведению анализа метаболома и интерактома, а также поиска путей таргетного лечения синдрома Ашера
Введение
Синдром Ашера (Usher syndrome, USH) является аутосомно-рецессивным заболеванием, на которое приходится почти половина всех случаев сочетанной наследственной глухоты и слепоты. По оценкам, распространенность USH составляет от 3,2 до 6,2 на 100 000 [1]. Тем не менее распространенность может быть до 1 на 6000. USH проявляется нарушением слуха, вестибулярной дисфункцией и пигментным ретинитом (RP) (синоним — тапеторетинальная абиотрофия сетчатки). В зависимости от возраста начала и тяжести клинических проявлений USH подразделяют на три основных клинических подтипа: USh2, USh3 и USh4. USh2 является наиболее тяжелой формой, наблюдающейся с рождения или с манифестацией до 5 лет, которая возникает в результате мутаций в любом из 6 генов: MYO7A, USh2C, CDh33, PCDh25, USh2G и CIB2. USh3 связан с мутациями в 3 генах: USh3A, ADGRV1 и DFNB31. USh4 вызван мутациями в гене CLRN1. Недавно был выделен 4-й атипичный USH, который возникает в результате мутаций в любом из генов: HARS, CEP250, PDZD7, C2orf71. На долю USh2 приходится около трети всех зарегистрированных случаев USH, на долю USh3 — две трети.
По оценкам, распространенность USH составляет от 3,2 до 6,2 на 100 000 [1]. Тем не менее распространенность может быть до 1 на 6000. USH проявляется нарушением слуха, вестибулярной дисфункцией и пигментным ретинитом (RP) (синоним — тапеторетинальная абиотрофия сетчатки). В зависимости от возраста начала и тяжести клинических проявлений USH подразделяют на три основных клинических подтипа: USh2, USh3 и USh4. USh2 является наиболее тяжелой формой, наблюдающейся с рождения или с манифестацией до 5 лет, которая возникает в результате мутаций в любом из 6 генов: MYO7A, USh2C, CDh33, PCDh25, USh2G и CIB2. USh3 связан с мутациями в 3 генах: USh3A, ADGRV1 и DFNB31. USh4 вызван мутациями в гене CLRN1. Недавно был выделен 4-й атипичный USH, который возникает в результате мутаций в любом из генов: HARS, CEP250, PDZD7, C2orf71. На долю USh2 приходится около трети всех зарегистрированных случаев USH, на долю USh3 — две трети. USh4 протекает клинически наиболее благоприятно, встречается очень редко (~3%).
USh4 протекает клинически наиболее благоприятно, встречается очень редко (~3%).
В течение последних нескольких лет были предприняты значительные усилия для описаний молекулярных характеристик, спектра мутаций и диагностики USH в различных популяциях, в т. ч. в испанской [2], американской [3], европейской [1], датской [4], финской [5], британской [1], алжирской [6], итальянской [7], немецкой [8], арабской [9], африканской [6] и китайской [10]. Современные методы секвенирования следующего поколения (next generation sequencing — NGS) значительно улучшили эффективность молекулярной характеристики в USH [1, 3, 11–13]. Кроме того, традиционный метод Сэнгера и метод мультиплексной амплификации (Multiplex ligation-dependent probe amplification — MLPA) успешно применяются для выявления глубоких интронных мутаций, больших делеций/дупликаций и вариаций числа копий генов (copy number variation — CNV).
Работы, в которых были представлены молекулярный профиль пациентов при центральных дистрофиях сетчатки в российской популяции [14] и генотип/фенотип корреляции в единичных клинических случаях пациентов с USH [15], уже были опубликованы авторами. Тем не менее нет опубликованных данных по USH в российской когорте в соответствии с утвержденным протоколом клинических исследований.
Тем не менее нет опубликованных данных по USH в российской когорте в соответствии с утвержденным протоколом клинических исследований.
Цель исследования: изучить спектр мутаций российской когорты пациентов с USH в рамках наблюдательного исследования NCT03319524 [16] и провести анализ метаболома и интерактома, а также патогенетических путей развития USH для поиска путей таргетного лечения.
Материал и методы
28 пациентов из группы 3214 пациентов (11 мужчин, 17 женщин; возраст — 47,6±12,7 года; возрастной диапазон проявления заболевания — 0–18 лет) с наиболее выраженными клиническими симптомами USH (нейросенсорная тугоухость 3–4 степени и сужение полей зрения до 5–15 градусов) были приглашены к участию. Национальный состав группы: 21 пациент (75%) — русские, 3 (10%) — украинцы, 2 (7%) — евреи, 1 (4%) — белорус и 1 (4%) — чуваш. Письменное информированное согласие было получено у всех 28 пациентов, исследование было одобрено Независимым международным этическим комитетом (Москва).
Клиническое обследование: каждому пациенту проводилась биомикроскопия, периметрия, визометрия, оптическая когерентная томография (ОКТ), офтальмоскопия с фоторегистрацией, электроретинография (ЭРГ), запись зрительно вызванных потенциалов, 32-оттеночный цветотест Хью, тональная и электронная аудиометрия, измерение акустического импеданса, вестибулометрия, электронистагмография и постурометрия. Молекулярно-генетическое подтверждение клинического диагноза выполнено методом высокопроизводительного параллельного секвенирования ДНК. Был проведен подробный компьютерный анализ патогенетических путей развития клинической картины у 20 пациентов с подтвержденным диагнозом USH. Использовалось программное обеспечение HGVS, Genemap, Genemania, CLINVAR, Genego, STRING, Cytoscape и база данных консорциума EMBL для моделирования интерактома и анализа полученных данных.
Результаты и обсуждение
Частота обнаруженных мутаций в обследованной когорте пациентов составила: 50% в гене USh3A, 39% — MYO7A, 5% — PCDh25, 3% — USh2C, 3% — CDh33, характеристика которых представлена авторами ранее [17] (табл. 1). Для обсуждения терапевтических подходов к коррекции утраченных или недостаточных функций генов в данной группе пациентов необходимо изучение и сопоставление всех факторов, которые влияют на патогенез заболевания, в т. ч. учет молекулярно-генетических факторов и факторов внешней среды: (геном —> транскриптом —> протеом —> метаболом), в совокупности образующих интерактом, которые определяют фенотип того или иного пациента [18]. Регуляции метаболизма в сетчатке обеспечиваются:
1). Для обсуждения терапевтических подходов к коррекции утраченных или недостаточных функций генов в данной группе пациентов необходимо изучение и сопоставление всех факторов, которые влияют на патогенез заболевания, в т. ч. учет молекулярно-генетических факторов и факторов внешней среды: (геном —> транскриптом —> протеом —> метаболом), в совокупности образующих интерактом, которые определяют фенотип того или иного пациента [18]. Регуляции метаболизма в сетчатке обеспечиваются:
1) количеством ферментов в зависимости от активации или ингибирования транскрипции и синтеза белков; 2) каталитической активностью ферментов, что контролируется аллостерическими модуляторами, активаторами, конкурентными ингибиторами (т. е. агонистами и антагонистами) и посттрансляционными модификациями, такими как фосфорилирование, ацетилирование и гликозилирование под контролем гормонов, ростовых факторов и нейротрансмиттеров; 3) субстратной доступностью (концентрацией), которая зависит от систем активного мембранного транспорта (помпы, транспортеры, требующие затрат энергии) и пассивной диффузии через субстрат-специфические мембранные белки (ионные каналы), которые контролируются концентрационным градиентом.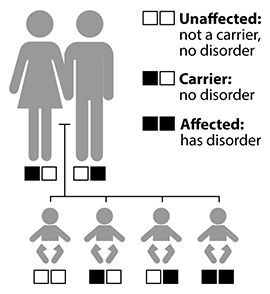 В процессе фототрансдукции происходит передача зрительного сигнала, начиная с захвата фотонов сетчаткой глаза и завершая формированием зрительных образов в зрительной коре головного мозга. Подробнее о геномном контроле фототрансдукции можно узнать, изучая функцию каждого из белков фототрансдукции, транслируемых с указанных генов, сначала по отдельности, а затем собирая в одну общую сеть интерактома. Изучение интерактома, т. е. полного набора взаимодействий между молекулами в отдельной клетке, как непосредственных физических контактов между белками (белок-белковые взаимодействия), так и непрямых взаимодействий генов (например, эпистаз, ап-регулирование, ко-активация, влияние факторов транскрипции и пр.), позволяет выделять наиболее перспективные терапевтические мишени. В то время как данные об экспрессии матричной рибонуклеиновой кислоты (мРНК) генов и данные протеомного анализа не раскрывают полностью всего того, что может происходить в клетке, метаболические профили (изучение метаболома сетчатки) [19, 20] могут дать мгновенный снимок физиологических процессов в клетке.
В процессе фототрансдукции происходит передача зрительного сигнала, начиная с захвата фотонов сетчаткой глаза и завершая формированием зрительных образов в зрительной коре головного мозга. Подробнее о геномном контроле фототрансдукции можно узнать, изучая функцию каждого из белков фототрансдукции, транслируемых с указанных генов, сначала по отдельности, а затем собирая в одну общую сеть интерактома. Изучение интерактома, т. е. полного набора взаимодействий между молекулами в отдельной клетке, как непосредственных физических контактов между белками (белок-белковые взаимодействия), так и непрямых взаимодействий генов (например, эпистаз, ап-регулирование, ко-активация, влияние факторов транскрипции и пр.), позволяет выделять наиболее перспективные терапевтические мишени. В то время как данные об экспрессии матричной рибонуклеиновой кислоты (мРНК) генов и данные протеомного анализа не раскрывают полностью всего того, что может происходить в клетке, метаболические профили (изучение метаболома сетчатки) [19, 20] могут дать мгновенный снимок физиологических процессов в клетке. Одна из задач современной медицины, системной биологии и функциональной геномики — это интегрирование данных протеомики, транскриптомики и метаболической информации для получения более целостного представления о строении и функции сетчатки у конкретного пациента. При компьютерном анализе и изучении взаимодействия наиболее активных генов метаболизма сетчатки при USH в изучаемой когорте выяснилось, что в 67% случаев наблюдается физическое взаимодействие генов, в 13% — ко-экспрессия, в 6% — ко-локализация, в 4% — совместные метаболические пути, в 1,4% — генные взаимодействия, 0,59% имеют общие белковые домены. В 6% случаев взаимодействие генов или белков, синтезируемых с этих генов, не доказано, но по некоторым предсказательным алгоритмам их взаимодействие вероятно.
Одна из задач современной медицины, системной биологии и функциональной геномики — это интегрирование данных протеомики, транскриптомики и метаболической информации для получения более целостного представления о строении и функции сетчатки у конкретного пациента. При компьютерном анализе и изучении взаимодействия наиболее активных генов метаболизма сетчатки при USH в изучаемой когорте выяснилось, что в 67% случаев наблюдается физическое взаимодействие генов, в 13% — ко-экспрессия, в 6% — ко-локализация, в 4% — совместные метаболические пути, в 1,4% — генные взаимодействия, 0,59% имеют общие белковые домены. В 6% случаев взаимодействие генов или белков, синтезируемых с этих генов, не доказано, но по некоторым предсказательным алгоритмам их взаимодействие вероятно.
Анализ интерактома (рис. 1) показывает, что у протокадгерина (PCDh25) большая сеть контактов с кадгеринами (CDh33), у ушерина (USh3A) — с разными видами коллагенов (COL4A3), у миозина (MYO7A) — с вирлиновым комплексом (WHRN) и различными подвидами актина (ACTA2, ACTG2). Примечательно, что MYO7A взаимодействует с гармонином, что позволяет предположить, что он может участвовать в закреплении верхнего конца цилии стержня фоторецептора (видоизмененные реснички). MYO7A также взаимодействует с некоторыми сплайс-формами протокадгерина (PCDh25), предполагается, что этот моторный белок может участвовать в формировании комплексов на обоих концах реснички (рис. 2).
Примечательно, что MYO7A взаимодействует с гармонином, что позволяет предположить, что он может участвовать в закреплении верхнего конца цилии стержня фоторецептора (видоизмененные реснички). MYO7A также взаимодействует с некоторыми сплайс-формами протокадгерина (PCDh25), предполагается, что этот моторный белок может участвовать в формировании комплексов на обоих концах реснички (рис. 2).
MYO7A — ген, кодирует одну из форм белка миозина, расположен на 11-й хромосоме и имеет 55 экзонов. Миозин действует как моторный белок, управляемый АТФазой, для транспорта меланосом и фагосом вдоль нитей актина в пигментном эпителии сетчатки, а также для транспорта опсина и других белков фототрансдукции в фоторецепторах. Мутации в гене MYO7A вызывают тяжелую и глубокую непрогрессирующую нейросенсорную тугоухость, пигментный ретинит до начала подросткового периода и часто с вестибулярной арефлексией. Острота центрального зрения обычно лучше, чем 0,3, в первые два десятилетия жизни. Поля зрения у пациентов с USh2B начинают сужаться в средней периферии и переходят к остаточным небольшим центральным островкам с небольшими участками периферических полей зрения на самых поздних стадиях заболевания. Офтальмоскопически у пациентов наблюдаются стушеванность артерий и атрофия ретинального пигментного эпителия (РПЭ) с пигментацией по типу «костных телец». Аутофлуоресценция глазного дна может показать кольцо гипераутофлуоресценции в макуле. ОКТ часто выявляет потерю наружных слоев сетчатки и присутствие кистозного макулярного отека. Ганцфельд-ЭРГ почти всегда нерегистрируемая или амплитуды сигналов сильно снижены.
Поля зрения у пациентов с USh2B начинают сужаться в средней периферии и переходят к остаточным небольшим центральным островкам с небольшими участками периферических полей зрения на самых поздних стадиях заболевания. Офтальмоскопически у пациентов наблюдаются стушеванность артерий и атрофия ретинального пигментного эпителия (РПЭ) с пигментацией по типу «костных телец». Аутофлуоресценция глазного дна может показать кольцо гипераутофлуоресценции в макуле. ОКТ часто выявляет потерю наружных слоев сетчатки и присутствие кистозного макулярного отека. Ганцфельд-ЭРГ почти всегда нерегистрируемая или амплитуды сигналов сильно снижены.
В сетчатке большая часть молекул миозина MYO7A находится в пигментном эпителии сетчатки (РПЭ), где происходят многие реакции зрительного ретиноидного цикла. V.S. Lopes et al. наблюдали [21], что сетчатка с мутациями в MYO7A устойчива к острому световому повреждению по причине более низкого уровня RPE65, изомеразы RPE, которая играет ключевую роль в ретиноидном цикле. Было показано, что RPE65 обычно подвергается светозависимой транслокации, чтобы стать более сконцентрированным в центральной области клеток RPE. Эта транслокация требует участия моторного белка MYO7A, поэтому при мутациях в гене MYO7A RPE65 частично локализован на свету и распадается быстрее, возможно, из-за его неправильной локализации, что дает правдоподобное объяснение его более низким уровням. После 50–60% фотозасвета сетчатка с мутацией в MYO7A демонстрирует повышенные уровни всех трансретиниловых эфиров на начальных стадиях темнового восстановления, что согласуется с дефицитом активности RPE65. Наконец, MYO7A и RPE65 совместно иммунопреципитируются из клеточного лизата RPE антителами против любого из белков, и два белка были локализованы в одних и тех же участках клетки, что указывает на их прямое или косвенное взаимодействие. Вместе результаты подтверждают роль MYO7A в транслокации RPE65, иллюстрируя участие молекулярного мотора в пространственно-временной организации ретиноидного цикла в процессе фототрансдукции [21].
Было показано, что RPE65 обычно подвергается светозависимой транслокации, чтобы стать более сконцентрированным в центральной области клеток RPE. Эта транслокация требует участия моторного белка MYO7A, поэтому при мутациях в гене MYO7A RPE65 частично локализован на свету и распадается быстрее, возможно, из-за его неправильной локализации, что дает правдоподобное объяснение его более низким уровням. После 50–60% фотозасвета сетчатка с мутацией в MYO7A демонстрирует повышенные уровни всех трансретиниловых эфиров на начальных стадиях темнового восстановления, что согласуется с дефицитом активности RPE65. Наконец, MYO7A и RPE65 совместно иммунопреципитируются из клеточного лизата RPE антителами против любого из белков, и два белка были локализованы в одних и тех же участках клетки, что указывает на их прямое или косвенное взаимодействие. Вместе результаты подтверждают роль MYO7A в транслокации RPE65, иллюстрируя участие молекулярного мотора в пространственно-временной организации ретиноидного цикла в процессе фототрансдукции [21].
На основе этих знаний можно предположить, что зарегистрированный и разрешенный к применению на территории США препарат Luxturna (воретиген непарвовек), представляющий собой рабочую копию гена RPE65 на аденовирусном носителе, в будущем может показать свою эффективность при терапии MYO7A, ассоциированного с USh2. Также разрабатывается генно-терапевтический препарат UshStat (компания Sanofi совместно с Oxford biomedica) с доставкой правильно работающей копии гена MYO7A на лентивирусном носителе [22–25], однако продвижения в этой области происходят относительно медленно, в т. ч. в связи с повышенной иммуногенностью лентивирусной технологии доставки.
USh3A — ген, кодирующий белок ушерин, расположен на 1-й хромосоме и имеет 72 экзона. Ушерин содержит участки ламинина эндотелиального ростового фактора, домен пентаксина и множество участков фибронектина типа III, он находится в базальной мембране и важен для развития и поддержания гомеостаза внутреннего уха и сетчатки. Для этого гена было найдено несколько вариантов транскриптов, кодирующих разные изоформы белка. Мутации в гене USh3A являются наиболее частой причиной аутосомно-рецессивного несиндромального пигментного ретинита и USh3 в мировой статистике, однако в нашей группе пациентов в связи с особенностями формирования когорты (по критериям включения в группу принимались пациенты с наиболее серьезными нарушениями) большой процент составили пациенты с USh2. Никталопия и потеря периферического поля зрения в течение первых двух десятилетий, прогрессирующая с возрастом, наблюдались у пациентов с этим типом заболевания, хотя темпы прогрессирования варьировались с существенной внутри- и межсемейной фенотипической изменчивостью. Терапевтические подходы к коррекции недостаточной функции белка ушерина в связи с большим размером гена затруднены, однако разрабатываются различные подходы к доставке более длинных генов (так называемые наночастицы) и попытки замены частей гена (генная терапия мутаций в 13-м экзоне гена USh3A, разрабатываемая компанией ProQR), в т.
Для этого гена было найдено несколько вариантов транскриптов, кодирующих разные изоформы белка. Мутации в гене USh3A являются наиболее частой причиной аутосомно-рецессивного несиндромального пигментного ретинита и USh3 в мировой статистике, однако в нашей группе пациентов в связи с особенностями формирования когорты (по критериям включения в группу принимались пациенты с наиболее серьезными нарушениями) большой процент составили пациенты с USh2. Никталопия и потеря периферического поля зрения в течение первых двух десятилетий, прогрессирующая с возрастом, наблюдались у пациентов с этим типом заболевания, хотя темпы прогрессирования варьировались с существенной внутри- и межсемейной фенотипической изменчивостью. Терапевтические подходы к коррекции недостаточной функции белка ушерина в связи с большим размером гена затруднены, однако разрабатываются различные подходы к доставке более длинных генов (так называемые наночастицы) и попытки замены частей гена (генная терапия мутаций в 13-м экзоне гена USh3A, разрабатываемая компанией ProQR), в т. ч. с применением технологии CRISPR [26]. Описание исследованной российской когорты пациентов с синдромом Ашера и частота мутаций в генах, наиболее частых при данном синдроме, представлены на рисунке 3 и в таблице 1.
ч. с применением технологии CRISPR [26]. Описание исследованной российской когорты пациентов с синдромом Ашера и частота мутаций в генах, наиболее частых при данном синдроме, представлены на рисунке 3 и в таблице 1.
PCDh25 — ген суперсемейства кадгеринов, кодирует один из подтипов протокадгерина, расположен на 10-й хромосоме и насчитывает 48 экзонов. Протокадгерины кодируют интегральные мембранные белки, которые обеспечивают кальций-зависимую клеточно-клеточную адгезию. Он играет существенную роль в поддержании нормальной функции сетчатки и кортиева органа. CDh33 вместе с PCDh25 образуют концевые звенья, которые соединяют стереоцилии, что крайне важно для поддержания жизнеспособности фоторецепторов и клеток кортиева органа. Мутации в этом гене приводят к потере слуха и развитию USh2. Характерен альтернативный сплайсинг, что приводит к появлению нескольких изоформ белка [27].
USh2C — ген ушерин 1С, расположен на 11-й хромосоме, имеет 29 экзонов. Этот ген кодирует белок-каркас, который участвует в сборке белковых комплексов. Белок содержит PDZ-домены, область сверхскрученной спирали с двойным сигналом ядерной локализации. Дефекты в этом гене являются причиной развития USh2C и несиндромальной нейросенсорной тугоухости аутосомно-рецессивного типа 18. Для этого гена было обнаружено множество вариантов транскриптов, кодирующих разные изоформы белка.
Этот ген кодирует белок-каркас, который участвует в сборке белковых комплексов. Белок содержит PDZ-домены, область сверхскрученной спирали с двойным сигналом ядерной локализации. Дефекты в этом гене являются причиной развития USh2C и несиндромальной нейросенсорной тугоухости аутосомно-рецессивного типа 18. Для этого гена было обнаружено множество вариантов транскриптов, кодирующих разные изоформы белка.
CDh33 — кадгерин-связанный белок 23-го подтипа, расположен на 10-й хромосоме, имеет 71 экзон. Этот ген также является членом суперсемейства кадгеринов. Белок с этого гена участвует в организации стереоцилий и формировании ресничек фоторецепторов. USh2D и несиндромальная аутосомно-рецессивная глухота DFNB12 вызваны аллельными мутациями этого кадгерин-подобного гена. Описаны альтернативные варианты сплайсинга, кодирующие разные изоформы этого белка [28].
Терапевтические подходы к коррекции функции генов USh2C, CDh33 и PCDh25 пока ограничиваются косвенными методиками — использованием аминогликозидов как ex vivo, так и in vivo для считывания преждевременных стоп-кодонов или нонсенс-мутаций [29]. Аминогликозиды в данном случае работают путем ослабления строгого механизма отбора рибосомой при синтезе белка из мРНК. Как при антибактериальном действии, когда они нарушают правильную генерацию полипептидов бактерий, так и в этом случае, расслабляя трансляцию, аминогликозид позволяет вставлять аминокислоту в область стоп-кодона вместо обрыва синтеза белка. Хотя эта аминокислота может быть не той аминокислотой, которая наблюдается в белке дикого типа, существует возможность восстановления правильной функции или по крайней мере лучшая функциональность получаемого белка ушерина, кадгерина или протокадгерина.
Аминогликозиды в данном случае работают путем ослабления строгого механизма отбора рибосомой при синтезе белка из мРНК. Как при антибактериальном действии, когда они нарушают правильную генерацию полипептидов бактерий, так и в этом случае, расслабляя трансляцию, аминогликозид позволяет вставлять аминокислоту в область стоп-кодона вместо обрыва синтеза белка. Хотя эта аминокислота может быть не той аминокислотой, которая наблюдается в белке дикого типа, существует возможность восстановления правильной функции или по крайней мере лучшая функциональность получаемого белка ушерина, кадгерина или протокадгерина.
Заключение
Авторами изложены результаты наблюдательного клинического исследования NCT03319524 и проведен подробный компьютерный анализ метаболома и интерактома, а также патогенетических путей развития USH в обследуемой когорте пациентов с обсуждением важности патогенетически направленных подходов к терапии и наиболее перспективных подходов к терапевтическому воздействию, в т. ч. с применением генной терапии.
ч. с применением генной терапии.
Финансирование/Funding
Исследование спонсировано БФ «Фонд поддержки слепоглухих «Со-единение» и АНО «Лаборатория Сенсор-Тех».
Autonomous non-commercial organization Laboratory “Sensor technology for deafblind” and Deaf-Blind Support Foundation “Con-nection”.
Сведения об авторах:
1Иванова Марианна Евгеньевна — к.м.н., руководитель, ORCID iD 0000-0002-1089-4293;
2Атарщиков Дмитрий Сергеевич — к.м.н., врач-офтальмолог, ORCID ID 0000-0003-4401-9099;
3Демчинский Андрей Михайлович — к.м.н., руководитель медицинских проектов, ORCID iD 0000-0002-1689-9394;
4Стрельников Владимир Викторович — д.б.н., заведующий лабораторией эпигенетики, ORCID iD 0000-0001-9283-902X;
5Бар Дебмала — PhD, начальник отдела исследований
и разработки, ORCID iD 0000-0002-2557-7768;
6Порядин Геннадий Васильевич — д. м.н., профессор, член-корр. РАН, почетный заведующий кафедрой патофизиологии, ORCID iD 0000-0003-2010-3296;
м.н., профессор, член-корр. РАН, почетный заведующий кафедрой патофизиологии, ORCID iD 0000-0003-2010-3296;
7Балашова Лариса Маратовна — д.м.н., профессор, руководитель, ORCID iD 0000-0001-9349-7092;
6Салмаси Жеан Мустафаевич — д.м.н., профессор, заведующий кафедрой патофизиологии, ORCID iD 0000-0001-8524-0019.
1НКЦ «Офтальмик», 125167, Россия, г. Москва, Ленинградский пр-т, д. 47/3–3.
2ФГБУ «ЦКБ с поликлиникой». 121359, Россия, г. Москва, ул. Маршала Тимошенко, д. 15.
3АНО «Лаборатория Сенсор-Тех». 115114, Россия, г. Москва, Павелецкая наб., д. 2, стр. 3.
4ФГБНУ “МГНЦ”. 115478, Россия, г. Москва, ул. Москворечье, д. 1.
5Центр геномики и прикладной генной технологии. 560032, Индия, г. Бангалор, ул. Чоланаяканахалли, д. 209.
6ФГБОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России. 117513, Россия, г. Москва, ул. Островитянова, д. 1.
Н.И. Пирогова Минздрава России. 117513, Россия, г. Москва, ул. Островитянова, д. 1.
7НП «МНПЦПТ». 119034, Россия, г. Москва, ул. Пречистенка, д. 29/14.
Контактная информация: Иванова Марианна Евгеньевна, e-mail: [email protected]. Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах. Конфликт интересов: Иванова М.Е. является сотрудником НКЦ «Офтальмик». Остальные авторы заявляют об отсутствии конфликта интересов. Статья поступила 07.05.2019.
About the authors:
1Marianna E. Ivanova — MD, PhD, Head of CRO, ORCID iD 0000-0002-1089-4293;
2Dmitry S. Atarshchikov — MD, PhD, ophthalmologist, ORCID iD 0000-0003-4401-9099;
3Andrey M. Demchinsky— MD, PhD, Head of Medical Projects, ORCID iD 0000-0002-1689-9394;
4Vladimir V. Strelnikov — PhD, Head of Epigenetics Laboratory, ORCID iD 0000-0001-9283-902X;
Strelnikov — PhD, Head of Epigenetics Laboratory, ORCID iD 0000-0001-9283-902X;
5Debmalya Barh — PhD, Head of R&D Department, ORCID iD 0000-0002-2557-7768;
6Gennadiy V. Poryadin — MD, PhD, Professor, Corresponding Member of RAS, Honorary Head of Pathophysiology Department, ORCID iD 0000-0003-2010-3296;
7Larisa M. Balashova — MD, PhD, Professor, Head of the Center, ORCID iD 0000-0001-9349-7092;
6Jean M. Salmasi — MD, PhD, Professor, Head of Pathophysiology Department, ORCID iD 0000-0001-8524-0019.
1LLC “Oftalmic”. 47/3–3, Leningradsky Prospekt, Moscow,125167, Russian Federation.
2Central Clinical Hospital for Presidential Affairs. 15, Marshala Timoshenko str., Moscow,121359, Russian Federation.
3Autonomous nonprofit organization “Scientific and industrial laboratory “Sensor technology for deafblind”.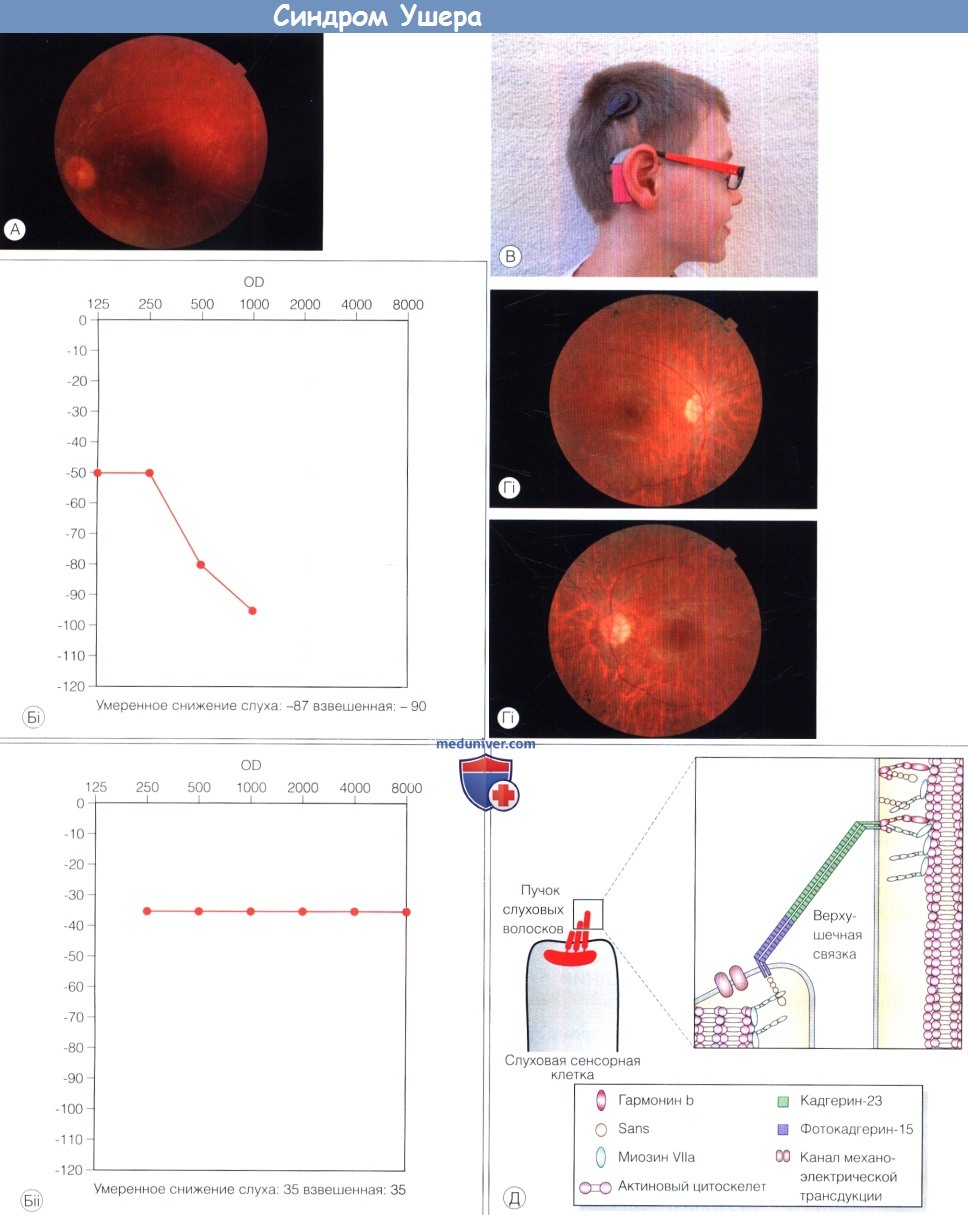 2, bld. 3, Paveletskaya naberezhnaya, Moscow, 115114, Russian Federation.
2, bld. 3, Paveletskaya naberezhnaya, Moscow, 115114, Russian Federation.
4Research Centre for Medical Genetics. 1, Moskvorechie str., Moscow, 115478, Russian Federation.
5Institute of Integrative Omics and Applied Biotechnology. 209, Cholanayakanahalli str., Bangalore, 560032, India.
6Pirogov Russian National Research Medical University. 1, Ostrovityanov str., Moscow, 117513, Russian Federation.
7Non-profit partnership International Scientific and Practical Center for the Proliferation of Tissues of Russia. 29/14, Prechistenka str., Moscow, 119034, Russian Federation.
Contact information: Marianna E. Ivanova, e-mail: [email protected]. Financial Disclosure: Marianna E. Ivanova is the collaborator of LLC “Oftalmic”. Other authors declare that there is no conflict of interests. Received 07.05.2019.
Usher Syndrome Coalition
Usher синдром является наиболее распространенной генетической причиной комбинированной глухоты и слепоты. Синдром Usher (USH) — это редкое наследственное заболевание, передающееся от родителей к детям, которое влияет на три основных чувства в организме: зрение, слух и равновесие .
Существует три клинических типа синдрома Ушера: Тип 1 обычно вызывает глубокую глухоту при рождении, нарушение вестибулярного аппарата (равновесия) и прогрессирующую потерю зрения; Тип 2 обычно вызывает умеренную или тяжелую потерю слуха при рождении и прогрессирующую потерю зрения; Тип 3 обычно вызывает прогрессирующую потерю слуха и зрения с более поздним началом. По оценкам, от него страдают не менее 25 000 человек в США и более 400 000 человек во всем мире.
В настоящее время нет лекарства от синдрома Ашера, но существует растущее сообщество USH. Построение сообщества ведет к лечению и излечению.
Построение сообщества ведет к лечению и излечению.
Коалиция Синдром Ашера — единственная организация в мире, работающая над поиском и поддержкой каждого человека и семьи, живущих с USH, независимо от того, где они живут, с каким типом USH они родились или как они общаются. Наша миссия — повышать осведомленность и ускорять исследования, предоставляя информацию и поддержку людям и семьям, страдающим синдромом Ашера. Мы стремимся быть наиболее полным ресурсом для сообщества людей с синдромом Ашера, сокращая разрыв между исследователями и семьями. Узнайте больше и примите участие.
Коалиция по борьбе с синдромом Ашера: объединение глобального сообщества Ашеров.
Вместе мы сможем сделать синдром Ашера историей.
Блог и новости о синдроме Ашера
4 мая 2023 г.
Endogena Therapeutics: исследование повышения дозы для оценки безопасности/переносимости и эффективности EA-2353 у субъектов с ретинитом
Летом 2022 года Endogena запустила первую дозу фазы 1/2a исследования EA-2353 при пигментном ретините (RP).
4 мая 2023 г.
Оценка качества сна и утомляемости у пациентов с синдромом Ушера 2а типа
В недавней публикации в Ophthalmology Science исследователи из Radboudumc стремились изучить значение проблем со сном и усталости, испытываемых пациентами с синдромом Usher типа 2a (USh3a).
1 мая 2023 г.
Препараты, повышающие устойчивость к стрессу, сохраняют структуру и функцию ткани в дегенерирующей сетчатке за счет ингибирования фосфодиэстеразы.
Прогрессирующие заболевания сетчатки, такие как возрастная дегенерация желтого пятна (AMD) и пигментный ретинит, вызваны дегенерацией сетчатки из-за апоптоза или гибели клеток из-за клеточного стресса.
Читать далее
OHSU подтверждает первую модель синдрома Ушера у нечеловеческих приматов
Генетические мутации приводят к тому, что люди с синдромом Ушера рождаются глухими, испытывают проблемы с равновесием и постепенно теряют зрение. Исследователи OHSU теперь смогут применить на практике способность корректировать экспрессию генов, ответственных за синдром Ашера. Usher поражает примерно от 4 до 17 из каждых 100 000 человек. (Getty Images)
Исследователи OHSU теперь смогут применить на практике способность корректировать экспрессию генов, ответственных за синдром Ашера. Usher поражает примерно от 4 до 17 из каждых 100 000 человек. (Getty Images)
У людей с синдромом Ашера — основной наследственной причиной одновременной глухоты и слепоты, от которой нет лечения — может появиться новая причина для надежды, теперь, когда исследователи из Орегонского университета здоровья и науки подтвердили первое когда-либо нечеловеческий примат модели их болезни. Подтверждение означает, что исследовательская группа, наконец, может протестировать генную терапию этого состояния и потенциально помочь больным детям сохранить зрение.
Генетические мутации приводят к тому, что люди с синдромом Ашера рождаются глухими, испытывают проблемы с равновесием и постепенно теряют зрение. Лечение Ашера, которым страдают примерно от 4 до 17 из каждых 100 000 человек, зашло в тупик из-за отсутствия модели на животных, которая точно имитирует то, как болезнь поражает людей.
Исследовательская группа OHSU работала над устранением этого пробела. Они подтвердили, что их модель, макака-резус, родившаяся год назад, имеет симптомы, которые отражают наиболее тяжелую форму синдрома Ашера, тип 1B. Команда сообщила об этих результатах во время презентации 11 февраля на собрании Ассоциации исследований в области отоларингологии. Исследователи использовали технологию редактирования генов CRISPR/Cas9.создать модель и тем самым сделать возможным тестирование экспериментальной генной терапии синдрома Ашера.
Марта Нойрингер, доктор философии. (OHSU)
«Хотя дети с Usher 1B рождаются глухими, кохлеарные имплантаты могут дать им хороший слух, особенно если они имплантированы достаточно рано», — сказала руководитель исследовательской группы, Марта Нойрингер, доктор философии . , профессор неврологии в Орегонском национальном исследовательском центре приматов OHSU и доцент-исследователь офтальмологии в Медицинской школе OHSU.
«Однако в настоящее время нет лечения, чтобы остановить неуклонно увеличивающуюся потерю зрения, которая происходит у детей с Usher 1B», — сказал Нойрингер. «Вот почему так важно иметь точную модель Usher. Мы надеемся и стремимся к тому, чтобы эта модель однажды позволила нам сохранить зрение у детей с синдромом Ушера».
Презентация на собрании была представлена членом команды Джоном В. Бригандом, доктором философии. , главный исследователь Орегонского исследовательского центра слуха OHSU и профессор отоларингологии/хирургии головы и шеи в Медицинской школе OHSU.
Джон Бриганд, доктор философии. (OHSU)
«Создание этой модели — поистине важное научное достижение, — сказал Бриганд. «Это нужно кричать с Эвереста».
Такие ученые, как Бриганд, уже используют мышей для изучения потери слуха по Ашеру, но фундаментальные различия в анатомии глаз означают, что мыши не являются подходящей моделью для потери зрения по Ашеру. Недавно была создана свиная модель другой формы болезни, Usher Type 1C.
Но поскольку глаза и зрение нечеловеческих приматов и людей почти идентичны, нечеловеческие приматы лучше всего помогают ученым понять заболевания сетчатки человека и оценить возможные методы лечения. Однако синдром Ашера в природе не встречается у нечеловеческих приматов. Поэтому Бриганде, Нойрингеру и их коллегам пришлось генетически сконструировать нечеловеческого примата с мутацией гена, вызывающей Ашера.
В команду разработчиков модели входят специалисты в области генетики и репродукции из отдела репродуктивных наук и исследований развития приматов. Они использовали технологию редактирования генов CRISPR/Cas9, чтобы вставить мутацию в ген MYO7A, которая вызывает Usher Type 1B у эмбрионов обезьян. Эмбрионы были перенесены суррогатным матерям-обезьянам для создания беременности.
В результате в конце 2021 года родился первый младенец с полным редактированием гена MYO7A. Тестирование быстро подтвердило, что у новорожденного макаки-резуса не было функционального слуха, а его ген MYO7A был мутирован. Он также показал нарушение равновесия, что привело к шаткой, неровной походке. Но поскольку потеря зрения у Ашера происходит постепенно, исследовательской группе пришлось подождать еще некоторое время. Когда макаке было 4 месяца, ученые начали замечать признаки того, что ее сетчатка — ткань в задней части глаза, которая обеспечивает зрение — начала ухудшаться, и эти изменения ухудшились в течение первого года.
Он также показал нарушение равновесия, что привело к шаткой, неровной походке. Но поскольку потеря зрения у Ашера происходит постепенно, исследовательской группе пришлось подождать еще некоторое время. Когда макаке было 4 месяца, ученые начали замечать признаки того, что ее сетчатка — ткань в задней части глаза, которая обеспечивает зрение — начала ухудшаться, и эти изменения ухудшились в течение первого года.
Теперь, когда команда подтвердила, что их модель имеет все три определяющих признака синдрома Ушера, они сосредоточили свое внимание на разработке экспериментальной генной терапии, предназначенной для доставки нормального гена MYO7A в сетчатку для противодействия сетчатке. вырождение. Их работа по генной терапии продолжается, и команда ожидает получить ранние результаты, чтобы поделиться ими позже в этом году.
Неизвестно, может ли эта модель также помочь разработать варианты лечения глухоты, вызванной синдромом Ашера. Люди с этим заболеванием рождаются с такими глубокими нарушениями слуха, что эксперты подозревают, что момент рождения может быть уже слишком поздним. Однако предыдущее исследование Brigande показало, что фетальная терапия или лечение, проводимое внутриутробно, может быть другим вариантом.
Однако предыдущее исследование Brigande показало, что фетальная терапия или лечение, проводимое внутриутробно, может быть другим вариантом.
Это исследование поддерживается Фондом борьбы со слепотой и Национальным институтом здравоохранения (гранты P51 OD011092, R21 DC018126).
Все исследования с участием животных в OHSU должны быть рассмотрены и одобрены университетским комитетом по уходу и использованию животных (IACUC) . Приоритетом IACUC является обеспечение здоровья и безопасности субъектов исследований на животных. IACUC также рассматривает процедуры, обеспечивающие здоровье и безопасность людей, работающих с животными. IACUC проводит тщательную проверку всех предложений по исследованиям на животных, чтобы убедиться, что они оправдывают использование живых животных и выбранных видов; наметить шаги, чтобы свести к минимуму боль и дистресс; задокументировать соответствующую подготовку всего задействованного персонала; и установить путем подробного обзора опубликованных источников, что предлагаемое исследование не дублирует без необходимости предыдущее исследование.