Митохондриальный синдром: Митохондриальные заболевания, комплексная диагностика: митохондриальная ДНК, ч. м. (Mitochondrial Diseases, multiplex mutations detection assay)

Почечные признаки первичных митохондриальных заболеваний
Резюме. Проведен обзор подходов к выявлению митохондриальной нефропатии в клинической практике
В зависимости от фенотипа различают синдромальные и несиндромальные формы первичных митохондриальных заболеваний (ПМЗ). Первые характеризуются сходной «мозаикой» симптомов и признаков, имеющих общую причину, а также вовлеченностью в патологический процесс сразу нескольких органов или типов тканей. Синдром полиорганного митохондриального заболевания может быть определяем с момента появления первых клинических признаков или развиться в ходе болезни. Наиболее часто ПМЗ поражают мышечную систему, периферические нервы, центральную нервную систему, органы зрения и слуха, эндокринную систему, сердце, легкие, желудочно-кишечный тракт, почки, кожу.
При ПМЗ почки вовлечены в патологический процесс в значительной степени. Однако до сих пор не существовало системного обзора этого вопроса. Ученые из Венского муниципального госпиталя (Municipal Hospital Rudolfstiftung), Австрия, и Федерального университета Сан-Паулу (Federal University of São Paulo), Бразилия, в совместной работе стремятся восполнить этот пробел. Системный обзор опубликован в апреле 2017 г. на страницах журнала «Biomedical Reports». Его целью является обобщение и обсуждение предшествующих выводов касательно почечных проявлений ПМЗ.
Системный обзор опубликован в апреле 2017 г. на страницах журнала «Biomedical Reports». Его целью является обобщение и обсуждение предшествующих выводов касательно почечных проявлений ПМЗ.
Поскольку ПМЗ чаще всего присутствует в виде синдрома полиорганного митохондриального заболевания, почечные признаки обычно включают: почечную недостаточность, нефролитиаз, нефротический синдром, кисты почек, почечный канальцевый ацидоз (ПКА), Барттер-подобный синдром, синдром Фанкони, фокальный сегментарный гломерулосклероз, тубулоинтерстициальный нефрит, нефрокальциноз, доброкачественные или злокачественные новообразования.
Среди синдромальных ПМЗ поражение почек наиболее часто выявляли у пациентов с митохондриальной энцефаломиопатией, молочнокислым ацидозом с инсультоподобными эпизодами, синдромом Кернса — Сейра, синдромом Ли и синдромами митохондриального истощения. Единичные случаи: сочетание почечной митохондриальной патологии с хронической прогрессирующей внешней офтальмоплегией; синдромом Пирсона; наследственной оптической нейропатией Лебера; дефицитом коэнзима Q10; Х-хромосомной сидеробластной анемией и атаксией; миопатией; молочным ацидозом; собственно сидеробластной анемией; дефицитом пируватдегидрогеназы; задержкой физического развития; аминоацидурией; холестазом; перегрузкой железом; лактатацидозом и ранней смертью; гиперурикемией; легочной гипертензией; почечной недостаточностью в младенчестве и алкалоз-синдромом. Авторы предполагают, что частота поражения почек при ПМЗ, по оценкам клиницистов, вероятно занижена. В диагностике поражений почек врачи следуют общим рекомендациям и зачастую проводят симптоматическое лечение. Важность почечных проявлений при ПМЗ, определяющих исход болезни, требует дополнительного признания и надлежащего контроля.
Авторы предполагают, что частота поражения почек при ПМЗ, по оценкам клиницистов, вероятно занижена. В диагностике поражений почек врачи следуют общим рекомендациям и зачастую проводят симптоматическое лечение. Важность почечных проявлений при ПМЗ, определяющих исход болезни, требует дополнительного признания и надлежащего контроля.
Митохондриальные нефропатии бывают первичными и вторичными (в результате первичного поражения других органов). Так, причиной инфаркта почки может стать фибрилляция предсердий или сердечная недостаточность; возможны поражения почек в результате первичной митохондриальной артериальной гипертензии; при митохондриальном диабете почечная недостаточность может быть следствием первичного поражения поджелудочной железы; причиной почечной недостаточности может стать рабдомиолиз, митохондриальная миопатия или митохондриальная эпилепсия.
Первичные почечные признаки ПМЗ включают в себя острую или хроническую почечную недостаточность, нефролитиаз, нефротический синдром, кисты почек, ПКА, Барттер-подобный синдром, синдром де Тони — Дебре — Фанкони, фокальный сегментарный гломерулосклероз, тубулоинтерстициальный нефрит, нефрокальциноз и доброкачественные или злокачественные новообразования.
Степень вовлечения почек в ПМЗ может быть классифицирована по количеству дополнительно затронутых органов. В отсутствие синдрома полиорганного митохондриального заболевания весьма сложно заподозрить именно митохондриальную природу болезни почек. Кроме того, существует классификация по фенотипу с разным уровнем доминантности. Дальнейшая дифференцировка поражений почек при ПМЗ зависит от клинических или субклинических проявлений. Для исследования субклинического уровня требуется биопсия или аутопсия почки с целью выявления морфологических аномалий митохондрий.
Диагноз ПМЗ основывается на результатах анализа крови, мочи, общего осмотра, функциональных тестов и биопсии. Помимо нарушений митохондриальной морфологии, биопсия может выявить дегенеративные изменения эпителиальных клеток почечных канальцев. Дополнительные биохимические исследования помогают выявить дисфункцию одного или нескольких комплексов митохондриальной дыхательной цепи; генетические — мутации в митохондриальной ДНК. У одного пациента может быть выявлено сразу несколько видов почечной патологии.
У одного пациента может быть выявлено сразу несколько видов почечной патологии.
Лечение митохондриальной нефропатии такое же, как при немитохондриальной нефропатии, однако следует избегать применения цитотоксичных препаратов, способных повлиять на функции митохондрий. Отмечен позитивный результат поддерживающего лечения антиоксидантами, кофакторами и витаминами. Пациентам со стероидрезистентным нефротическим синдромом требуется прием коэнзима Q10. В соответствии с тяжестью митохондриальной почечной недостаточности может потребоваться применение гемодиализа.
Результаты выполненного обзора свидетельствуют, что поражение почек при ПМЗ возникает чаще, чем первоначально предполагалось, а первичное и вторичное вовлечение почек в патологический процесс возможно как при синдромальных, так и при несиндромальных ПМЗ.
Диагностика поражений почек при ПМЗ аналогична стандартной диагностике заболеваний этого органа, но для пациентов с Барттер-подобным синдромом необходимо исключить собственно синдром Барттера. Если митохондриальная нефропатия является начальным проявлением ПМЗ, то для адекватной диагностики требуется проведение биопсии.
Если митохондриальная нефропатия является начальным проявлением ПМЗ, то для адекватной диагностики требуется проведение биопсии.
Александр Гузий
Митохондриальный оксидативный стресс и метаболический синдром
Современная эпидемиология метаболического синдрома в значительной степени определяется чрезмерным потреблением питательных веществ и отсутствием физической активности. Тем не менее, метаболический синдром является не только следствием взаимодействия основных причин, но и включает в себя множественные метаболические и физиологические изменения.
Метаболический синдром и митохондриальный оксидативный стресс.
Метаболический синдром обычно включает в себя все или некоторые из перечисленных состояния: резистентность к инсулину, ожирение, неалкогольный жировой гепатоз печени, гипертонию, сердечно-сосудистые заболевания, про-воспалительный статус в результате накопления жировой ткани. Хотя метаболический синдром возникает из-за сложного взаимодействия между генетическими и экологическими факторами, ясно, что основными факторами риска являются хроническое переедание и физическая неактивность. Поэтому наиболее значимой и понятной причиной нынешней эпидемии метаболического синдрома является тот факт, что человечество развивалось в условиях постоянной физической активности и переменной доступности пищи, и, следовательно, человеческий организм имеет возможность сохранять избыток энергии во времена изобилия. И теперь мы живем в среде, где пища всегда доступна, а физическая активность отсутствует. Однако, на биохимическом уровне механизмы, с помощью которых хроническое чрезмерное потребление пищи и отсутствие физической активности приводят к развития метаболического синдрома сложны и не полностью понятны. Отчасти, это связано с тем, что развитие фенотипа метаболического синдрома включает взаимодействие между печенью, поджелудочной железой, скелетными мышцами и жировой тканью, а также, их регуляцией и координацией гипоталамическими нейронами мозга. Несмотря на это, диапазон данных многих исследования показывает, что усиление митохондриального оксидативного стресса часто ассоциируется и может способствовать развитию метаболического синдрома.
Поэтому наиболее значимой и понятной причиной нынешней эпидемии метаболического синдрома является тот факт, что человечество развивалось в условиях постоянной физической активности и переменной доступности пищи, и, следовательно, человеческий организм имеет возможность сохранять избыток энергии во времена изобилия. И теперь мы живем в среде, где пища всегда доступна, а физическая активность отсутствует. Однако, на биохимическом уровне механизмы, с помощью которых хроническое чрезмерное потребление пищи и отсутствие физической активности приводят к развития метаболического синдрома сложны и не полностью понятны. Отчасти, это связано с тем, что развитие фенотипа метаболического синдрома включает взаимодействие между печенью, поджелудочной железой, скелетными мышцами и жировой тканью, а также, их регуляцией и координацией гипоталамическими нейронами мозга. Несмотря на это, диапазон данных многих исследования показывает, что усиление митохондриального оксидативного стресса часто ассоциируется и может способствовать развитию метаболического синдрома. Определение роли митохондрий в развитии МС выглядит привлекательно, поскольку изменения физической активности и избыток питательных веществ, которые предрасполагают к метаболического синдрома, воздействуют непосредственно на функцию митохондрий и значительно влияют на выработку митохондриями активных форм кислорода (АФК).
Определение роли митохондрий в развитии МС выглядит привлекательно, поскольку изменения физической активности и избыток питательных веществ, которые предрасполагают к метаболического синдрома, воздействуют непосредственно на функцию митохондрий и значительно влияют на выработку митохондриями активных форм кислорода (АФК).
Поступление топлива в митохондрии при переедании.
Митохондрии преобразуют энергию, запасённую в питательных веществах, в соотношение АТФ/АДФ, которое обеспечивает работу тела и служит источником АТФ для большинства клеток организма. Серия механизмов организованных по принципу обратной связи регулирует скорость митохондриального оксидативного фосфорилирования в зависимости от потребности клеток в АТФ. Если поступление углеводов превышает потребность для синтеза АТФ, они буду экспортироваться из митохондрий в виде цитрата и превращаться в цитозоле в жирные кислоты, для хранения в виде жира. Различные сигнальные пути, прежде всего инсулиновый путь, объединяют то, как организм в целом накапливает энергию при избытке питания. Упрощенная схема выглядит следующим образом: после еды повышение уровня глюкозы в плазме повышает уровень инсулина, который действуя через инсулиновый рецептор способствует перемещению траспортера глюкозы GLUT4 на поверхность плазматической мембраны скелетных мышц для преобразования в жир и использования в будущих энергетических процессах. Затем избыток глюкозы поглощается жировыми тканями в ответ на действие инсулина и превращается в жир для хранения в виде триглицеридов, а также хранения в печени в виде гликогена. Однако, чрезмерное питание и недостаточная физическая активность приводят к инсулинорезистентности и накоплению жира в жировой ткани, печени и мышцах. Сосредоточимся на метаболической ситуации в скелетных мышцах, поскольку эта ткань имеет переменную потребность в АТФ, и, следовательно, поглощение и накопление углеводов и жиров быстро изменяется в ответ на диетические и гормональные сигналы, особенно в ответ на действие инсулина. Следовательно, эта система охватывает основные механизмы которыми пересекаются митохондриальные функции и метаболический синдром.
Упрощенная схема выглядит следующим образом: после еды повышение уровня глюкозы в плазме повышает уровень инсулина, который действуя через инсулиновый рецептор способствует перемещению траспортера глюкозы GLUT4 на поверхность плазматической мембраны скелетных мышц для преобразования в жир и использования в будущих энергетических процессах. Затем избыток глюкозы поглощается жировыми тканями в ответ на действие инсулина и превращается в жир для хранения в виде триглицеридов, а также хранения в печени в виде гликогена. Однако, чрезмерное питание и недостаточная физическая активность приводят к инсулинорезистентности и накоплению жира в жировой ткани, печени и мышцах. Сосредоточимся на метаболической ситуации в скелетных мышцах, поскольку эта ткань имеет переменную потребность в АТФ, и, следовательно, поглощение и накопление углеводов и жиров быстро изменяется в ответ на диетические и гормональные сигналы, особенно в ответ на действие инсулина. Следовательно, эта система охватывает основные механизмы которыми пересекаются митохондриальные функции и метаболический синдром. И раскрытые принципы могут, в большей или меньшей степени, применяться к другим тканям.
И раскрытые принципы могут, в большей или меньшей степени, применяться к другим тканям.
Митохондрии как источник АФК при переедании и отсутствии физической активности.
Митохондриальная дыхательная цепь является основным источником АФК внутри клетки. Активная форма кислорода (O2—) генерируется митохондриями и является супероксидом, который сам по себе не является реактивным, но который может повреждать Fe-S связи в белках. Основная судьба супероксида в митохондриях – это быть быстро диспропорционированым до пероксида водорода в реакции первого порядка с помощью марганецзависимой супероксиддисмутазы (MnSOD), которая в больших количествах представлена в матриксе. Перекись водорода выpывает окислительное повреждение, особенно в присутствии металлов и тем самым разрушает липиды, белки и ДНК. Сокращение пула носителей электронов связанных с дыхательной цепью митохондрии (НАДН, флавины, убихинон), низкая скорость процессов тканевого дыхания, протон-движущая сила приводят к повышению концентрации кислорода в митохондриях и создают условия, благоприятствующие производству митохондриального супероксида. В митохондриях, которые активно производят АТФ, выработка супероксида снижена, поскольку носители электронов находятся в окисленном состоянии, скорость тканевого дыхания высокая. Напротив, переедание поставляет лишние электроны в дыхательную цепь, в то время как отсутствие физической активности, и как следствие, низкий спрос на АТФ, будут способствовать накоплению протонов в межмембранном пространстве и увеличению протон-движущей силы и образованию супероксида. Повышение продукции митохондриальных АФК может быть связано с патологией вызванной с метаболическим синдромом тремя путями. Это может быть вторичным следствием развития фенотипа метаболического синдрома, при котором митохондриальные АФК не способствуют непосредственно прогрессированию расстройства. Однако это маловерятно, поскольку в эксперименте на животных было показано, что вмешательства снижающие уровень АФК в митохондриях предотвращают развитие метаболического синдрома. Другой аспект заключается в том, что повышенный уровень АФК является следствием состояний, возникающих во время чрезмерного питания и отсутствия физической активности, что приводит к неспецифическому оксидативному повреждению митохондрий.
В митохондриях, которые активно производят АТФ, выработка супероксида снижена, поскольку носители электронов находятся в окисленном состоянии, скорость тканевого дыхания высокая. Напротив, переедание поставляет лишние электроны в дыхательную цепь, в то время как отсутствие физической активности, и как следствие, низкий спрос на АТФ, будут способствовать накоплению протонов в межмембранном пространстве и увеличению протон-движущей силы и образованию супероксида. Повышение продукции митохондриальных АФК может быть связано с патологией вызванной с метаболическим синдромом тремя путями. Это может быть вторичным следствием развития фенотипа метаболического синдрома, при котором митохондриальные АФК не способствуют непосредственно прогрессированию расстройства. Однако это маловерятно, поскольку в эксперименте на животных было показано, что вмешательства снижающие уровень АФК в митохондриях предотвращают развитие метаболического синдрома. Другой аспект заключается в том, что повышенный уровень АФК является следствием состояний, возникающих во время чрезмерного питания и отсутствия физической активности, что приводит к неспецифическому оксидативному повреждению митохондрий. Хоть это и возможно, уровни окислительного повреждения митохондрий и изменение удельной активности их компонентов, наблюдаемые при метаболическом синдроме, имеют тенденции быть относительно скромными. Поэтому можно полагать, что нарастание неспецифического оксидативного повреждения митохондрий вряд ли сыграет главную роль в развитии метаболического синдрома, хотя в экстремальных случаях это может способствовать развитию патологии. Предпочтительней считать, что митохондриальные АФК являются частью механизма окислительно-восстановительной сигнализации, который смягчает баланс между использованием и хранением энергии. Нарушение этого механизма при хроническом переедании и недостаток АТФ могут способствовать развитию фенотипа метаболического синдрома.
Хоть это и возможно, уровни окислительного повреждения митохондрий и изменение удельной активности их компонентов, наблюдаемые при метаболическом синдроме, имеют тенденции быть относительно скромными. Поэтому можно полагать, что нарастание неспецифического оксидативного повреждения митохондрий вряд ли сыграет главную роль в развитии метаболического синдрома, хотя в экстремальных случаях это может способствовать развитию патологии. Предпочтительней считать, что митохондриальные АФК являются частью механизма окислительно-восстановительной сигнализации, который смягчает баланс между использованием и хранением энергии. Нарушение этого механизма при хроническом переедании и недостаток АТФ могут способствовать развитию фенотипа метаболического синдрома.
Митохондриальная редокс (окислительно-восстановительная) регуляция при метаболическом синдроме.
Продукция АФК как побочного продукта тканевого дыхания позволяет митохондриям быть частью редокс-сигнализационной цепи. Это происходит когда усиливается окислительно-восстановительный процесс, такой как увеличение продукции АФК, усиливается и генерируется сигнал, который изменяет активность целевого белка, а когда редокс процесс ослабляется, сигнал инвертируется. Изменения производства АФК митохондриями в ответ на увеличение отношения НАДН/НАД+ вызвано увеличением концентрации субстрата (переедание) или отсутствием митохондриального синтеза АТФ (малоподвижный образ жизни) может использоваться как индикатор митохондриального статуса. Предполагается, что продуцирование митоходриальных АФК необходимо для передачи клетке редокс-сигнала, который обеспечивает обратную связь для регуляции метаболизма митохондрий, и что при метаболическом синдроме это связь нарушается и способствует развитию патологии. Чтобы понять, как это может произойти, обратим внимание, что дыхательная цепь генерирует супероксид, который затем превращается в пероксид водорода. Обе эти АФК могут действовать как редокс сигналы, но по-разному.
Это происходит когда усиливается окислительно-восстановительный процесс, такой как увеличение продукции АФК, усиливается и генерируется сигнал, который изменяет активность целевого белка, а когда редокс процесс ослабляется, сигнал инвертируется. Изменения производства АФК митохондриями в ответ на увеличение отношения НАДН/НАД+ вызвано увеличением концентрации субстрата (переедание) или отсутствием митохондриального синтеза АТФ (малоподвижный образ жизни) может использоваться как индикатор митохондриального статуса. Предполагается, что продуцирование митоходриальных АФК необходимо для передачи клетке редокс-сигнала, который обеспечивает обратную связь для регуляции метаболизма митохондрий, и что при метаболическом синдроме это связь нарушается и способствует развитию патологии. Чтобы понять, как это может произойти, обратим внимание, что дыхательная цепь генерирует супероксид, который затем превращается в пероксид водорода. Обе эти АФК могут действовать как редокс сигналы, но по-разному. Предполагается, что эти оба вида АФК участвуют в регуляции метаболизма митохондрий. Хотя супероксид является основной формой АФК, продуцируемой дыхательной цепью митохондрий, он на самом деле не является слишком реактивной молекулой и значительно взаимодействует только с несколькими биологически значимыми мишенями, в частности, железо-серными белками, оксидом азота и хинонами. Анион супероксида не является мембранопроницаемым веществом и его основная судьба быть превращенным в перекись водорода марганецзависимой супероксиддисмутазы. Таким образом, супероксид компартментализирован, а его равновесная концентрация определяется балансом между скоростью его образования и дисмутации. Появляется все больше информации, что сам супероксид может действовать как окислительно-восстановительный сигнализатор в митохондриях. В эксперименте на мышах было показано, что удаление марганецзависимой супероксиддисмутазы (MnSOD) приводит к смерти в течении нескольких дней. Поскольку MnSOD обычно рассматривается как важный антиоксидантный фермент, было предположено, что эта летальность связана с окислительным повреждением.
Предполагается, что эти оба вида АФК участвуют в регуляции метаболизма митохондрий. Хотя супероксид является основной формой АФК, продуцируемой дыхательной цепью митохондрий, он на самом деле не является слишком реактивной молекулой и значительно взаимодействует только с несколькими биологически значимыми мишенями, в частности, железо-серными белками, оксидом азота и хинонами. Анион супероксида не является мембранопроницаемым веществом и его основная судьба быть превращенным в перекись водорода марганецзависимой супероксиддисмутазы. Таким образом, супероксид компартментализирован, а его равновесная концентрация определяется балансом между скоростью его образования и дисмутации. Появляется все больше информации, что сам супероксид может действовать как окислительно-восстановительный сигнализатор в митохондриях. В эксперименте на мышах было показано, что удаление марганецзависимой супероксиддисмутазы (MnSOD) приводит к смерти в течении нескольких дней. Поскольку MnSOD обычно рассматривается как важный антиоксидантный фермент, было предположено, что эта летальность связана с окислительным повреждением. Но, тем не менее, мыши нокаутированные по MnSOD имеют нормальные уровни перекисного окисления липидов и не имеют повреждений митхондриальной ДНК, опровергая предположение, что окислительное повреждение вызывает преждевременную смерть. Вместо этого, мыши лишенные MnSOD страдают главным образом от нарушения обмена веществ с задержкой роста, гипотермией, гипертрофией сердца и накоплением жира в скелетных мышцах и печени. Это связывается с наблюдаемым 40% снижение активности аконитазы, железо-серного белка, который особенно чувствителен к действию супероксида. Можно сделать вывод, что основной функцией MnSOD есть предотвращение инактивации железосодержащих белков в митохондриальном матриксе, особенно аконитазы. Причина, по которой аконитаза особенно чувствительная к действию супероксида заключается в том, что этот фермент имеет в своём активном центре железо-серный центр, который необходим для его активности. Интересно, что реактивность аконитазы с супероксидом достаточно высокая даже в присутствии высоких концентраций MnSOD.
Но, тем не менее, мыши нокаутированные по MnSOD имеют нормальные уровни перекисного окисления липидов и не имеют повреждений митхондриальной ДНК, опровергая предположение, что окислительное повреждение вызывает преждевременную смерть. Вместо этого, мыши лишенные MnSOD страдают главным образом от нарушения обмена веществ с задержкой роста, гипотермией, гипертрофией сердца и накоплением жира в скелетных мышцах и печени. Это связывается с наблюдаемым 40% снижение активности аконитазы, железо-серного белка, который особенно чувствителен к действию супероксида. Можно сделать вывод, что основной функцией MnSOD есть предотвращение инактивации железосодержащих белков в митохондриальном матриксе, особенно аконитазы. Причина, по которой аконитаза особенно чувствительная к действию супероксида заключается в том, что этот фермент имеет в своём активном центре железо-серный центр, который необходим для его активности. Интересно, что реактивность аконитазы с супероксидом достаточно высокая даже в присутствии высоких концентраций MnSOD. Можно предположить, что аконитаза является не просто уязвимой мишенью окислительного повреждения супероксидом, а супероксид регулирует метаболизм в митоходриях посредством модулирования активности аконитазы. Следствием такого предположения является то, что необычайно высокая чувствительность аконитазы к супероксиду могла развиваться намеренно, в качестве регуляторной функции. Как инактивация аконитазы путем повышения уровня митохондриального супероксида может действовать как редокс-регуляторный мезханизм? Аконитаза является частью цикла Кребса и ее инактивация может привести к накоплению цитрата в матриксе. Цитрат является последним распространённый метаболит на пути полного окисления ацетил-КоА и его экспорта для синтеза жирных кислот в цитоплазме, пэоэтому инактивация аконитазы в ответ на митоходриальный супероксид может привести к исключению ацетил-КоА из процесса окислительного фосфолирирования и накоплению жира. Эта петля обратной связи также может быть частью механизма антиоксидатной защиты, предотвращая длительное образование супероксида.
Можно предположить, что аконитаза является не просто уязвимой мишенью окислительного повреждения супероксидом, а супероксид регулирует метаболизм в митоходриях посредством модулирования активности аконитазы. Следствием такого предположения является то, что необычайно высокая чувствительность аконитазы к супероксиду могла развиваться намеренно, в качестве регуляторной функции. Как инактивация аконитазы путем повышения уровня митохондриального супероксида может действовать как редокс-регуляторный мезханизм? Аконитаза является частью цикла Кребса и ее инактивация может привести к накоплению цитрата в матриксе. Цитрат является последним распространённый метаболит на пути полного окисления ацетил-КоА и его экспорта для синтеза жирных кислот в цитоплазме, пэоэтому инактивация аконитазы в ответ на митоходриальный супероксид может привести к исключению ацетил-КоА из процесса окислительного фосфолирирования и накоплению жира. Эта петля обратной связи также может быть частью механизма антиоксидатной защиты, предотвращая длительное образование супероксида. Это работает за счет замедления доставки носителей электронов, таких как НАДН в дыхательную цепь, тем самым уменьшая выработку АТФ. Кроме того, эта модель предполагает, что MnSOD играет значительную роль в регуляции энергетического обмена: при низкой активности MnSOD жирные кислоты легче синтезируются и сохраняются, в то время как при высокой активности MnSOD много энергии теряется в виде тепла при утечке протов через внутреннюю мембрану митохондрий. Несмотря на то, что в клетке есть несколько других, внемитохондриальных источников супероксида, драматические эффекты изменения экспрессии MnSOD на метаболизм и тот факт, что супероксид является мембрано-непроницаемым, делают маловероятной регулирующую роль других источников супероксида. Подводя итог, можно сделать вывод, что повышенная концентрация супероксида митоходрий, из-за избытка питательных веществ и/или отсутсвия потребности в АТФ, инактивирует аконитазу и тем самым переводит ацетил-КОА в митоходриальный матрикс на хранение в виде жира, а также действует как механизм обратной связи для уменьшения митоходриальной продукции АФК.
Это работает за счет замедления доставки носителей электронов, таких как НАДН в дыхательную цепь, тем самым уменьшая выработку АТФ. Кроме того, эта модель предполагает, что MnSOD играет значительную роль в регуляции энергетического обмена: при низкой активности MnSOD жирные кислоты легче синтезируются и сохраняются, в то время как при высокой активности MnSOD много энергии теряется в виде тепла при утечке протов через внутреннюю мембрану митохондрий. Несмотря на то, что в клетке есть несколько других, внемитохондриальных источников супероксида, драматические эффекты изменения экспрессии MnSOD на метаболизм и тот факт, что супероксид является мембрано-непроницаемым, делают маловероятной регулирующую роль других источников супероксида. Подводя итог, можно сделать вывод, что повышенная концентрация супероксида митоходрий, из-за избытка питательных веществ и/или отсутсвия потребности в АТФ, инактивирует аконитазу и тем самым переводит ацетил-КОА в митоходриальный матрикс на хранение в виде жира, а также действует как механизм обратной связи для уменьшения митоходриальной продукции АФК.
Выводы в отношении метаболического синдрома.
Предполагается, что существует механизм редокс-регуляции, который позволяет митохондриям ощущать и реагировать на чрезмерное питание и низкую потребность в АТФ, увеличивая образование супероксида и перекиси водорода. Эта регуляция позволяет митохондриям реагировать на чрезмерное питание замедлением цикла трикарбоновых кислот и β-окисления жирных кислот, одновременно увеличивая расщепление углеводов для накопления жира. Механизмы редокс-регуляции, с помощью которых митохондрии ощущают и реагируют на чрезмерное питание и отсутствие физической активности, могут объяснять почему при метаболическом синдроме повышаются уровни АФК. Митохондриальная продукция АФК может быть возможной терапевтической мишенью при борьбе с метаболическим синдромом, и это подтверждается несколькими исследованиями на животных, в которых антиоксиданты, нацеленные на митоходрии, продемонстрировали перспективу в борьбе с некоторыми аспектами метаболического синдрома. В частности, в будущем, возможно появится способ разработки фармакологических подходов для селективного воздействия на продукцию митохондриального супероксида и перекиси водорода, тем самым сводя к минимуму влияние чрезмерного питания и отсутствия физической активности на развитие метаболического синдрома.
В частности, в будущем, возможно появится способ разработки фармакологических подходов для селективного воздействия на продукцию митохондриального супероксида и перекиси водорода, тем самым сводя к минимуму влияние чрезмерного питания и отсутствия физической активности на развитие метаболического синдрома.
Централизованная городская
гормональная лаборатория
Врач лабораторной диагностики
В.И.Гончарук
Митохондриальная болезнь | UMDF
Что такое митохондриальная болезнь?
Когда митохондрии не могут преобразовывать пищу и кислород в энергию для поддержания жизни, происходит повреждение клеток и даже их гибель. Когда этот процесс повторяется во всем организме, системы органов начинают давать сбои и даже перестают функционировать.
Почему Митохондрии Неисправны?
Нам еще многое предстоит открыть, но это то, что мы знаем. Митохондриальная болезнь является наследственным заболеванием. На ваши митохондрии также могут влиять другие генетические нарушения и факторы окружающей среды.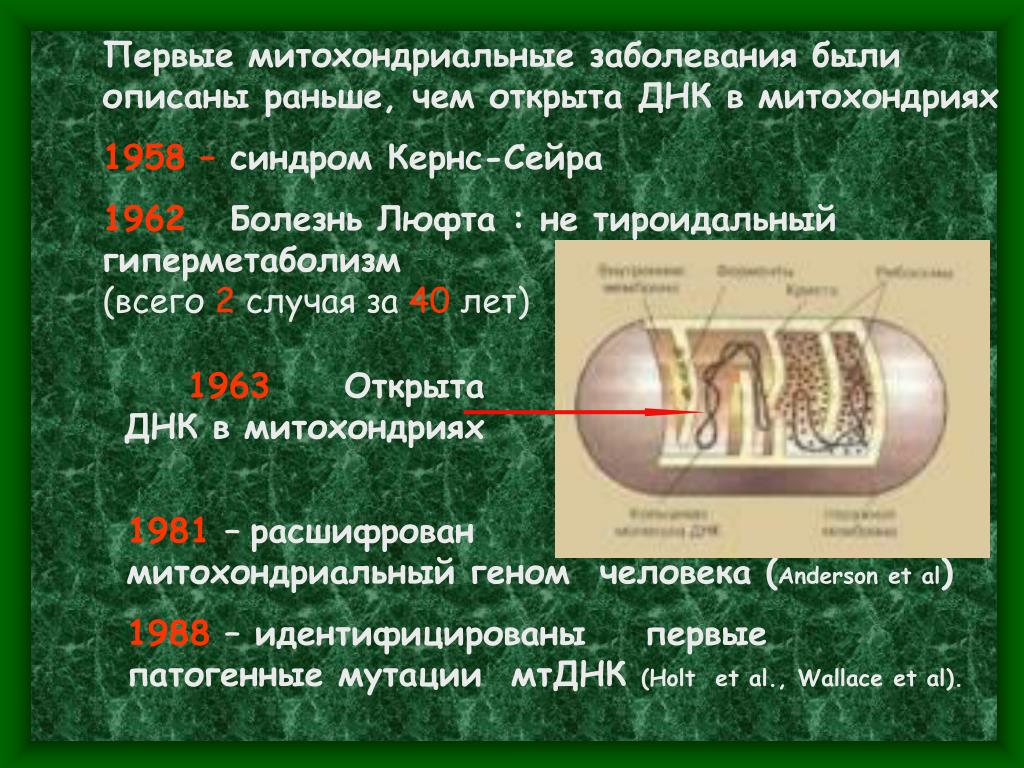 Вы можете узнать больше о биологии митохондриальных заболеваний здесь .
Вы можете узнать больше о биологии митохондриальных заболеваний здесь .
Каждые 30 минут рождается ребенок, у которого к 10 годам разовьется митохондриальное заболевание.
Как митохондриальное заболевание влияет на организм
наиболее подвержены митохондриальным заболеваниям. Пораженный человек может проявлять спектр симптомов.
задержка развития, деменция, мигрень, аутистические черты, судороги, инсульт, атипичный церебральный паралич, нарушение обучаемости
слабость/отказ, судороги, рефлюкс, рвота, запор, диарея, гипотония, нарушение моторики
обморок, отсутствие рефлексов, непереносимость тепла/холода, боль недостаточность почечной трубки
дефекты, закупорка, кардиомиопатия
низкий уровень сахара в крови, печеночная недостаточность
потеря зрения, птоз, атрофия зрительного нерва, косоглазие, офтальмоплегия, пигментный ретинит
потеря слуха
отсутствие набора веса, усталость, низкий рост, необъяснимая рвота, проблемы с дыханием
Узнайте, соответствуете ли вы требованиям для бесплатного генетического теста.
Начало работы →
Прочтите статью «Диагностика и лечение митохондриальных заболеваний».
Просмотреть документ →
UMDF ведет список более чем 200 врачей, лечащих и исследующих митохондриальные заболевания.
Поиск поставщика услуг →
Существует множество типов митохондриальных заболеваний. Каждое расстройство вызывает спектр аномалий, которые могут сбивать с толку как пациентов, так и врачей.
Узнать больше
3+ нарушение работы систем органов является тревожным сигналом для митохондриального заболевания.
Сколько людей страдают митохондриальной болезнью?
Точное число людей, страдающих митохондриальным заболеванием, определить трудно, потому что многим из них часто ставится неправильный диагноз. Один из 5000 человек имеет генетическое митохондриальное заболевание.
Могут ли взрослые болеть митохондриальными заболеваниями?
Хотя это заболевание в основном поражает детей, начало заболевания у взрослых становится все более распространенным.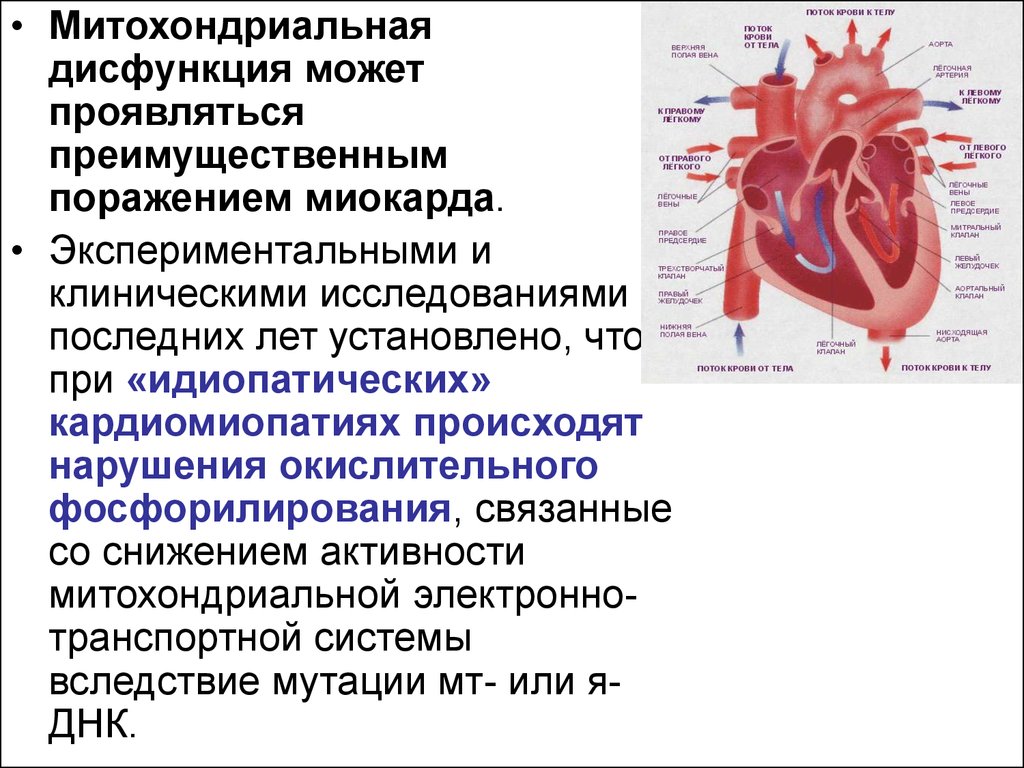 Сам процесс старения может быть результатом ухудшения функции митохондрий. Существует широкий спектр метаболических, наследственных и приобретенных нарушений у взрослых, которые можно отнести к аномальной функции митохондрий.
Сам процесс старения может быть результатом ухудшения функции митохондрий. Существует широкий спектр метаболических, наследственных и приобретенных нарушений у взрослых, которые можно отнести к аномальной функции митохондрий.
Когда человек с митохондриальным заболеванием относится к группе высокого риска?
Как дети, так и взрослые подвержены высокому риску неврологического и органного повреждения во время повышенных нагрузок на организм. Это включает в себя во время и в течение двух недель после болезни, голодание, обезвоживание, хирургическое вмешательство, анестезию и внутривенное введение антибиотиков.
Каков прогноз для лиц, страдающих митохондриальными заболеваниями?
На этот вопрос сложно ответить, поскольку прогноз зависит от множества критериев. Некоторые пострадавшие дети и взрослые живут вполне нормальной жизнью. В других случаях больные дети могут не дожить до подросткового возраста. Начало во взрослом возрасте может привести к резкому изменению образа жизни и физическим изменениям за короткий промежуток времени.
Как UMDF поддерживает исследования по поиску лекарства?
UMDF финансирует лучшую науку по всему миру, в том числе почти 12 миллионов долларов на исследования для улучшения методов лечения и лечения. Наша программа исследовательских грантов и 9Программа ускорителей 0022 поддерживает лучшие умы митохондриальной медицины.
Получите образование в Университете Мито
Получите доступ к видео, статьям и ресурсам, чтобы лучше ориентироваться в своем путешествии.
Посетите Mito U
В то время как мы боремся за поиск лекарства от митохондриального заболевания, существуют некоторые методы лечения, витамины и пищевые добавки, которые помогут облегчить симптомы и замедлить прогрессирование заболевания. Однако эффективность современных методов лечения сильно различается. Наша лучшая надежда на улучшение ухода за пациентами — это финансирование исследований митохондриальных заболеваний и клинических испытаний.
От 1000 до 4000 детей в США ежегодно рождаются с митохондриальными заболеваниями.
Присоединяйтесь к mitoSHARE: Реестр митохондриальных заболеваний
Помогите продвинуться вперед в направлении улучшения диагностики, лечения и лечения.
Зарегистрируйтесь
Зарегистрируйтесь в клиническом испытании, чтобы помочь в поиске лекарства
Узнайте, имеете ли вы право участвовать в клинических исследованиях.
Начало работы
Привлечение средств на исследования для поиска более эффективных методов лечения
Финансовые пожертвования программам поддержки UMDF для улучшения лечения митохондриальных заболеваний.
Подробнее
Типы митохондриальных заболеваний | УМДФ
ADOA — аутосомно-доминантная атрофия зрительного нерва ADOA — аутосомно-доминантная атрофия зрительного нерва — https://rarediseases.info.nih.gov/diseases/5243/autosomal-dominant-optic-atrophy-plus-syndrome DOA — доминантная атрофия зрительного нерва — DOA —. ..
подробнее
Болезнь или синдром Альперса Длинное название: Прогрессирующая детская полиодистрофия Симптомы: Судороги, деменция, спастичность, слепота, дисфункция печени и дегенерация головного мозга Источник: д-р Рольф Люфт; Развитие митохондриальной медицины. [Обзор]; Материалы…
[Обзор]; Материалы…
подробнее
Синдром Барта / LIC (летальная детская кардиомиопатия) Симптомы: Скелетная миопатия, кардиомиопатия, низкорослость и нейтропения Причина: X-сцепленный рецессивный источник: Dr. J. Christodoulou; Синдром Барта: клинические наблюдения и исследования генетического сцепления; Американский…
подробнее
Дефекты бета-окисления См. LCAD, LCHAD, MAD, MCAD, SCAD, SCHAD, VLCAD Лечение: диета с высоким содержанием углеводов и низким содержанием жиров, введение триглицеридного масла со средней длиной цепи и диетические добавки с карнитином и/или рибофлавином. Избегание голодания. ПРИМЕЧАНИЕ. Информация о…
подробнее
Дефицит карнитин-ацил-карнитинаСимптомы: судороги, апноэ, брадикардия, рвота, вялость, кома, увеличение печени, слабость в конечностях, миоглобин в моче, Рейе-подобные симптомы, вызванные голоданием Причина: аутосомно-рецессивный.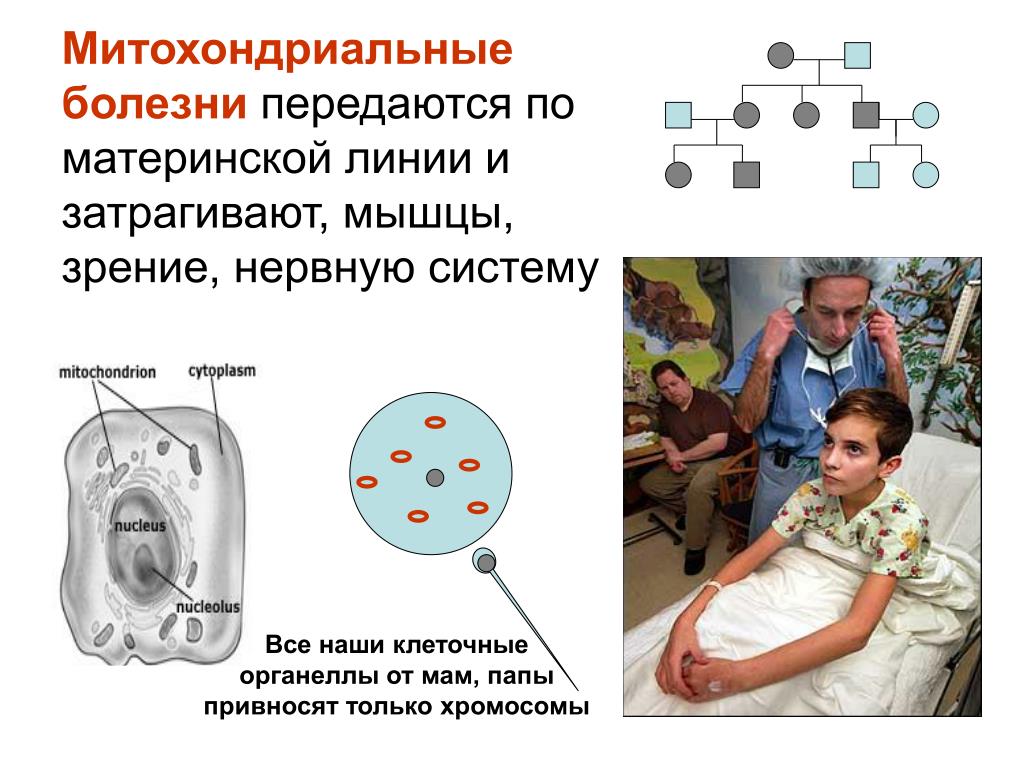 ..
..
читать далее
Дефицит карнитинаСимптомы: Судороги, апноэ, брадикардия, рвота, вялость, кома, увеличение печени, слабость конечностей, миоглобин в моче, Рейеподобные симптомы, вызванные голоданием Причина: Аутосомно-рецессивный…
подробнее
Дефицит комплекса I Длинное название: дефицит НАДН-дегидрогеназы (НАДН-CoQ редуктазы) Внутри митохондрии находится группа белков, которые переносят электроны по четырем цепным реакциям (комплексы I-IV), что приводит к выработке энергии. Эта сеть известна как Электрон…
читать далее
Комплекс II Дефицит Длинное название: Дефицит сукцинатдегидрогеназы Симптомы: Энцефаломиопатия и различные проявления, в том числе задержка развития, задержка развития, гиоптония, вялость, дыхательная недостаточность, атаксия, миоклонус. Часто встречается лактоацидоз. Может вызвать…
подробнее
Комплекс III Дефицит Длинное название: Дефицит убихинон-цитохром с оксидоредуктазы Симптомы: Четыре основные формы: фатальная детская энцефаломиопатия, врожденный лактоацидоз, гипотония, дистрофическая осанка, судороги и кома. Рвано-красные волокна обыкновенные….
Рвано-красные волокна обыкновенные….
подробнее
Дефицит комплекса IV / Дефицит ЦОГ Длинное название: Дефицит цитохром-с-оксидазы вызывается дефектом в комплексе IV дыхательной цепи. Симптомы: две основные формы: обычно нормальные в течение первых 6–12 месяцев жизни, затем наблюдается регресс развития…
подробнее
Дефицит комплекса V Длинное название: Дефицит АТФ-синтазы Симптомы: Медленно прогрессирующая миопатия.
Дефицит CPT I Симптомы: Увеличение печени и рецидивирующие Рейе-подобные эпизоды, вызванные голоданием или болезнями Причина: Аутосомно-рецессивное Лечение: Среднецепочечные триглицериды…
Подробнее
Дефицит коэнзима Q10Симптомы: энцефаломиопатия, умственная отсталость, непереносимость физической нагрузки, рваные красные волокна и рецидивирующий миоглобин в моче Причина: Вероятно, аутосомно-рецессивное заболевание Лечение: введение коэнзима Q10 Ссылки:. ..
..
Что такое CPEO?Смотрите похожие темы: KSS, Синдром делеции митохондрийХроническая прогрессирующая наружная офтальмоплегия (CPEO) представляет собой синдром делеции митохондриальной ДНК, характеризующийся слабостью глазных мышц. Заболевание обычно возникает у взрослых в возрасте…
подробнее
CPT II Симптомы дефицита – миопатические: непереносимость физической нагрузки, непереносимость голодания, мышечная боль, ригидность мышц и миоглобин в моче Симптомы – инфантильные: синдром Рея, увеличение печени, гипогликемия, увеличение сердца и сердечная аритмия Причина: аутосомно-доминантный.
подробнее
Синдромы дефицита креатина Дополнительные названия: синдромы дефицита церебрального креатина (CCDS) включают: дефицит гуанидиноацетатметилтрансферазы (дефицит GAMT), дефицит L-аргинин:глицинамидинотрансферазы (дефицит AGAT) и креатин, связанный с SLC6A8…
подробнее
Что такое KSS? См. также: CPEO, синдром делеции митохондрий Синдром Кернса-Сейра (KSS) — это синдром делеции митохондриальной ДНК, который поражает несколько систем организма. Это редкое заболевание, поражающее примерно 1,6 из 100 000 человек [1]. Самцы и…
также: CPEO, синдром делеции митохондрий Синдром Кернса-Сейра (KSS) — это синдром делеции митохондриальной ДНК, который поражает несколько систем организма. Это редкое заболевание, поражающее примерно 1,6 из 100 000 человек [1]. Самцы и…
подробнее
Молочный ацидоз Причина: Накопление молочной кислоты из-за превышения ее производства над потреблением. Хронический лактоацидоз является частым симптомом митохондриального заболевания. Ссылки: https://rarediseases.info.nih.gov/diseases/3163/lactic-acidosis-congenital-infantile 9.0005
подробнее
LBSL – ЛейкодистрофияЛейкоэнцефалопатия с поражением ствола головного и спинного мозга и повышением лактата (LBSL) является результатом мутации гена DARS2 и характеризуется медленно прогрессирующей мозжечковой атаксией и спастичностью с дисфункцией дорсального столба (снижение…
читать далее
LCAD Длинное название: дефицит длинноцепочечной ацил-КоА-дегидрогеназы. Симптомы: обычно вызывает смертельный синдром у младенцев, типичными признаками которого являются задержка развития, увеличение печени, увеличение сердца, метаболическая энцефалопатия и гипотония. Причина: Аутосомно-рецессивный. Лечение: См….
Симптомы: обычно вызывает смертельный синдром у младенцев, типичными признаками которого являются задержка развития, увеличение печени, увеличение сердца, метаболическая энцефалопатия и гипотония. Причина: Аутосомно-рецессивный. Лечение: См….
подробнее
LCHAD Симптомы: энцефалопатия, дисфункция печени, кардиомиопатия и миопатия. Также пигментная ретинопатия и периферическая невропатия. Причина: Аутосомно-рецессивный. Лечение: См. Дефекты бета-окисления. Ссылки:…
подробнее
Ежемесячные онлайн-встречи LHON Live Каждый месяц члены сообщества LHON собираются онлайн на LHON Live. Во время каждого сеанса пациенты общаются на темы, связанные с жизнью с LHON. Что такое ЛХОН? Наследственная оптическая нейропатия Лебера (LHON) является редким наследственным…
подробнее
Что такое синдром Ли? Синдром Лея (или болезнь Ли) представляет собой митохондриальное заболевание, иногда называемое подострой некротизирующей энцефаломиелопатией (СНЭ). Хотя это случается редко, эксперты считают его одним из наиболее распространенных клинических проявлений митохондриального заболевания[1]….
Хотя это случается редко, эксперты считают его одним из наиболее распространенных клинических проявлений митохондриального заболевания[1]….
читать дальше
Болезнь люфта. Симптомы: гиперметаболизм с лихорадкой, непереносимостью жары, обильным потоотделением, полифагией, полидипсией, рваными красными волокнами и тахикардией в покое. Непереносимость физических упражнений с легкой слабостью. Причина: неизвестное наследование NIH Link:…
подробнее
MAD / Глутаровая ацидурия Тип II Длинное название: Множественный дефицит ацил-КоА-дегидрогеназы Причина: Дефекты флавопротеинов, ответственных за перенос электронов (ETF или ETF-дегидрогеназы), что влияет на функцию всех шести ETF-концентраторов ацил-КоА…
подробнее
MCAD Длинное название: Дефицит ацил-КоА-дегидрогеназы средней цепи. Симптомы: поражает младенцев или детей младшего возраста с эпизодами энцефалопатии, увеличением и жировой дегенерацией печени и низким содержанием карнитина в крови. Причина: Аутосомно-рецессивный. Лечение: См….
Причина: Аутосомно-рецессивный. Лечение: См….
подробнее
MERRFLong Name: Миоклоническая эпилепсия и болезнь рваных красных волокон. Симптомы: миоклонус, эпилепсия, прогрессирующая атаксия, мышечная слабость и дегенерация, глухота и слабоумие.
подробнее
Что такое MELAS? Митохондриальная энцефаломиопатия с лактоацидозом и инсультоподобными эпизодами (MELAS) представляет собой прогрессирующее полисистемное заболевание, которое в первую очередь поражает нервную систему и мышцы. Несмотря на редкость, это одно из наиболее распространенных митохондриальных заболеваний.
подробнее
MEPAN Длинное название: митохондриальная нейродегенерация, связанная с белком еноил-КоА-редуктазы. MEPAN вызывается двумя мутациями в гене MECR (который кодирует белок митохондриальной транс-2-еноил-коэнзим-А-редуктазы), недавно описанном митохондриальном заболевании, которое.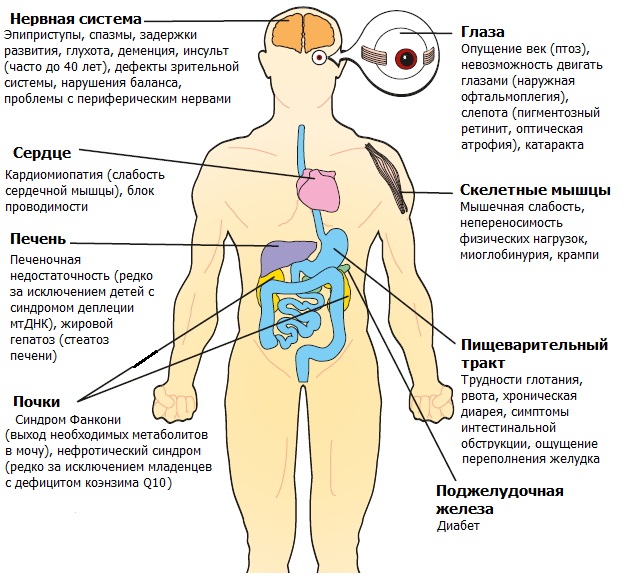 ..
..
подробнее
MIRASДлинное название: Синдром митохондриальной рецессивной атаксии. Симптомы: энцефалопатия, нарушения равновесия, атаксия, эпилепсия, когнитивные нарушения, психические симптомы, нарушения движения глаз, непроизвольные движения, периферическая невропатия. Причина: мутация POLG, рецессивный…
подробнее
Что такое делеционная болезнь митохондриальной ДНК? См. родственное заболевание: KSS, CPEO. Распространенная форма митохондриального заболевания возникает в результате отсутствия (или делеции) сегмента кольца митохондриальной ДНК (мтДНК). Как и большинство заболеваний мтДНК, эти нарушения обычно существуют в виде…
читать далее
Истощение митохондриальной ДНК. Симптомы: три формы: после нормального раннего развития до годовалого возраста появляется слабость, которая быстро ухудшается, вызывая дыхательную недостаточность и смерть, как правило, в течение нескольких лет. Неонатальная слабость, гипотония, требующая оказания помощи…
Неонатальная слабость, гипотония, требующая оказания помощи…
подробнее
Митохондриальная энцефалопатия Включает: энцефаломиопатию, энцефаломиелопатию.
MNGIE Длинное название: мионейрогастоинтестинальные расстройства и энцефалопатия. Симптомы: прогрессирующая наружная офтальмоплегия, слабость конечностей, периферическая невропатия, расстройства пищеварительного тракта, лейкодистрофия, лактоацидоз, рваные красные волокна. Новая ссылка NIH:…
подробнее
Длинное название NARP: нейропатия, атаксия и пигментный ретинит Причина: точечные мутации митохондриальной ДНК в генах, связанных с комплексом V: T8993G (также T8993C некоторыми исследователями). Синдром Лея может возникнуть, если процент мутации достаточно высок. Ссылки:…
подробнее
Синдром Пирсона Симптомы: дисфункция костного мозга и поджелудочной железы Причина: одиночные делеции митохондриальной ДНК. Наследование обычно спорадическое. У тех, кто выживает в младенчестве, обычно развивается синдром Кернса-Сейра. Ссылки:…
Наследование обычно спорадическое. У тех, кто выживает в младенчестве, обычно развивается синдром Кернса-Сейра. Ссылки:…
подробнее
Дефицит пируватдегидрогеназы Симптомы: молочнокислый ацидоз, атаксия, пировиноградный ацидоз, дегенерация позвоночника и мозжечка Реже: агенезия мозолистого тела и поражения базальных ганглиев, мозжечка и ствола головного мозга Также: задержка роста, гипотония, судороги,…
подробнее
Мутации
POLG См.: Митохондриальные новости, том 14, выпуск 2 См.: Заболевания, связанные с POLG – https://www.ncbi.nlm.nih.gov/pubmed/20301791 Ссылка NIH: https://ghr.nlm.nih.gov/gene/POLG
подробнее
Что такое первичные митохондриальные миопатии? Первичные митохондриальные миопатии (ПММ) представляют собой группу генетически определенных митохондриальных заболеваний, которые в основном поражают мышцы [1]. Эти расстройства могут поражать мужчин, женщин и людей любой этнической или расовой принадлежности. ..
..
читать далее
Дефицит пируваткарбоксилазы. Симптомы: лактоацидоз, гипогликемия, тяжелая задержка развития, задержка развития. Общие симптомы: судороги и спастичность. Причина: аутосомно-рецессивный.
подробнее
SANDO Ссылки: https://www.ncbi.nlm.nih.gov/books/NBK26471/
подробнее
SCAD Длинное название: Дефицит короткоцепочечной ацил-КоА-дегидрогеназы Симптомы: Задержка развития, задержка развития и гипогликемия Причина: Аутосомно-рецессивное Лечение: См. Дефекты бета-окисления Ссылки:…
Подробнее
SCHAD Симптомы: энцефалопатия и, возможно, заболевание печени или кардиомиопатия Причина: аутосомно-рецессивное заболевание Лечение: см. Дефекты бета-окисления Ссылки: https://rarediseases.info.nih.gov/diseases/9Дефицит 870/3-альфа-гидроксиацил-коа-дегидрогеназы
подробнее
Что такое TK2d? Дефицит тимидинкиназы 2 (TK2d) является редким генетическим митохондриальным заболеванием, которое также может называться дефектом поддержания митохондриальной ДНК, связанным с TK2, или синдромом истощения митохондриальной ДНК 2 (MTDPS2)[1].