
Митохондриальные болезни у детей: Митохондриальная патология у детей | #01/16

Актуальные вопросы лечения митохондриальных нарушений uMEDp
Среди ярких событий современной медицинской науки одно из значимых мест занимает появление области, которую все чаще называют «митохондриальной медициной». Она интересна со многих точек зрения. Во-первых, как и полагается новому систематическому объединению, она знаменует собой выделение новых патологических процессов и нозологических форм. Во-вторых, ее безусловное прикладное значение определяется наличием специфической, так называемой энерготропной, терапии.
Первичные и вторичные митохондриальные нарушения
Ключевая область этого раздела медицины – наследственные синдромы, в основе которых лежат мутации генов, ответственных за митохондриальные белки (синдромы Кернса – Сейра, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), MERRF (myoclonic epilepsy with ragged red fibers), Пирсона, Барта и др.). Однако класс состояний, характеризующихся митохондриальной недостаточностью, отнюдь не ограничивается этими «первичными» митохондриальными заболеваниями. Огромное количество болезней включает в себя нарушения клеточного энергообмена в качестве «вторичных» звеньев патогенеза: синдром хронической усталости, мигрени, кардиомиопатии, гликогенозы, заболевания соединительной ткани, диабет, рахит, тубулопатии, панцитопения, гипопаратиреоз, печеночная недостаточность и др. Особое значение для практической медицины имеет изучение указанных нарушений в связи с разработкой в этой области эффективных методов терапевтической коррекции.
К настоящему времени наиболее изучены дефекты, связанные с дефицитом различных комплексов дыхательной цепи и некоторых ферментов матрикса. Появляются клинические описания дефекта и других ферментов, например, наружной митохондриальной мембраны [1], так что до полного представления о тонких механизмах митохондриальной дисфункции еще далеко, и часто речь идет о недостаточности митохондриальной функции в целом. Ткани и органы зависят от митохондриальной активности в различной степени [2–5]. В их ряду на первом месте стоят нервные элементы, затем сердечная и скелетная мышечная ткани, почки, эндокринные железы и печень. Увеличение количества митохондрий и их структурные нарушения широко определяются в эндотелиальных клетках, гладких миоцитах и перицитах различных сосудов [6].
Ткани и органы зависят от митохондриальной активности в различной степени [2–5]. В их ряду на первом месте стоят нервные элементы, затем сердечная и скелетная мышечная ткани, почки, эндокринные железы и печень. Увеличение количества митохондрий и их структурные нарушения широко определяются в эндотелиальных клетках, гладких миоцитах и перицитах различных сосудов [6].
К болезням, причиной которых являются мутации митохондриальных генов, относятся синдромы Кернса – Сейра (нарушения со стороны глаз, атаксия, мышечная слабость, нарушения сердечной проводимости и другие симптомы), Пирсона (вялость, нарушения со стороны крови, кишечника, поджелудочной железы), MELAS (энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды), оптическая нейропатия Лебера и многие другие. Причем описание таких синдромов продолжается и сейчас – так, в XXI веке уже опубликованы описания нескольких новых заболеваний.
Поскольку все митохондрии достаются новому организму только от цитоплазмы яйцеклетки, многие митохондриальные заболевания являются или спорадическими, или наследуются с нарушением законов Менделя – «внеядерное» или «цитоплазматическое» наследование по материнской линии. Распространенность этих болезней плохо изучена, но ясно, что они относятся к сравнительно редким наследственным заболеваниям. Отсюда и малый интерес широкого круга медиков к митохондриальной патологии.
Однако эти заболевания, хотя и создали ядро «митохондриальной медицины», отнюдь не составляют всего ее спектра. В последние годы внимание медиков начал все больше приковывать следующий факт: несмотря на наличие в митохондриях собственной ДНК, кодируются ею всего около 2% белков, используемых в митохондриях. Иными словами, 98% наследственной информации о митохондриальных белках заложено в ядре, а значит, количество наследственных митохондриальных нарушений, связанных с ядерными мутациями, должно быть несоизмеримо больше тех, о которых упоминалось выше. А известно их на сегодняшний день не так много (среди них различные формы младенческих миопатий, болезни Альперса, Лея, Барта, Менкеса, синдромы недостаточности карнитина, некоторых ферментов цикла Кребса и дыхательной цепи), что понятно – маленькую митохондриальную ДНК гораздо легче изучать, чем гигантский ядерный геном. Таким образом, значительное число таких состояний сейчас можно предсказать только гипотетически.
Таким образом, значительное число таких состояний сейчас можно предсказать только гипотетически.
Интенсивное изучение признаков болезней клеточной энергетики приводит к еще более важному выводу: распространенность состояний, связанных с митохондриальной недостаточностью, не ограничивается наследственными синдромами, вызываемыми мутациями генов, непосредственно ответственных за митохондриальные белки. Умеренные нарушения клеточной энергетики могут не проявляться в виде самостоятельного заболевания, однако сказываются на характере течения других болезней. Широчайший круг других заболеваний включает в себя те или иные нарушения клеточной энергетики как вторичные звенья патогенеза.
Проведенные недавно в нашем институте исследования группы из 100 детей, поступивших в генетическую клинику с недифференцированными нарушениями физического и нервно-психического развития, показали, что у 49 из них отмечены нарушения клеточного энергообмена. Кроме того, нами выявлено влияние митохондриальной недостаточности на характер послеожогового рубцевания у детей, течение тонзиллитов, некоторых кардиологических, наследственных соединительнотканных, урологических и других заболеваний. Изучение этих патологических состояний и распространение информации о важности анализа энергетических дисфункций тем более актуально, что в настоящее время существуют действенные методы коррекции митохондриальной недостаточности, которые помогают в лечении перечисленных выше, не всегда истинно «митохондриальных» заболеваний.
Многие факторы окружающей среды и лекарственные препараты, вероятно, представляют собой существенную причину патологических изменений митохондрий. К этим факторам относится действие алкилирующих агентов (например, нитрозамины из окружающей среды), гидроксильных радикалов, высоких доз ультрафиолетового и ионизирующего излучений, лекарственных препаратов (бриостатин, азидотимидин), других химических агентов (аллоксан, цианиды, моноокись углерода и др.). Причиной митохондриального повреждения может быть и недостаточность некоторых микроэлементов, например селена.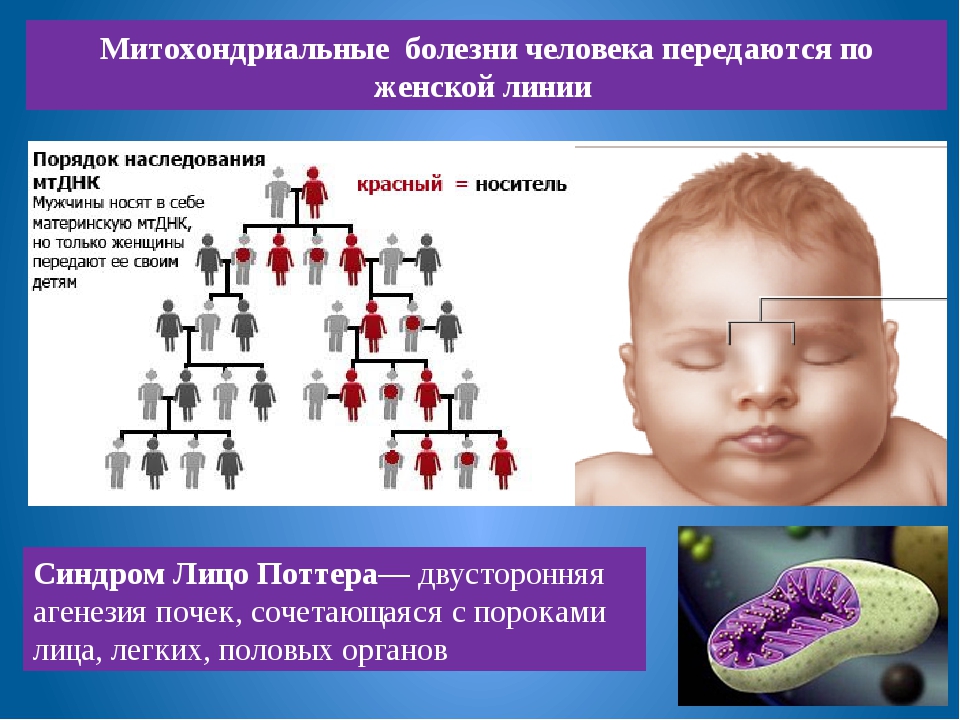 Во многих случаях чувствительность митохондриальной ДНК к действию повреждающих факторов окружающей среды в несколько раз выше по сравнению с чувствительностью ядерного генома. В целом область патогенетически целесообразного использования препаратов, воздействующих на митохондрии, включает в себя:
Во многих случаях чувствительность митохондриальной ДНК к действию повреждающих факторов окружающей среды в несколько раз выше по сравнению с чувствительностью ядерного генома. В целом область патогенетически целесообразного использования препаратов, воздействующих на митохондрии, включает в себя:
Лечение:
- митохондриальных болезней;
- «вторичных» (эндогенных и экзогенных) митохондриальных нарушений при других заболеваниях и состояниях.
Профилактика:
- возможных осложнений различных заболеваний у пациентов с энергодефицитным диатезом;
- преждевременных патологических нарушений, связанных с пожилым возрастом.
- Реабилитационные мероприятия при различных хронических заболеваниях.
Кроме того, целесообразно применение энерготропных препаратов в качестве стимуляторов адаптационных процессов при заболеваниях, не несущих митохондриальной дисфункции в качестве патогенетической составляющей. Эта область наименее изучена, однако, по нашим данным, повышенная митохондриальная пролиферация может обладать важным адаптационным потенциалом, компенсирующим функциональный дефект при некоторых заболеваниях (например, при врожденных структурных миопатиях).
Общая характеристика энерготропных препаратов
Потенциальные возможности лечения митохондриальных болезней распределяются по трем основным направлениям [7]:
- Применение фармакологических препаратов и биологически активных добавок.
- Модификация макронутриентной поддержки, диетотерапия.
- Использование реабилитационных методов лечебной физкультуры.
Терапевтические подходы к лечению митохондриальных болезней подразделяются на семь категорий [8]:
- паллиативная терапия;
- удаление вредных метаболитов;
- применение искусственных акцепторов электронов;
- применение метаболитов и кофакторов;
- применение поглотителей кислородных радикалов;
- генная терапия;
- генетическое консультирование.

Большая группа лекарственных препаратов, которые принято называть метаболическими, пользуется чрезвычайной популярностью у широкого круга врачей. Можно без преувеличения сказать, что лекарства и нелекарственные средства, в разных количествах и соотношениях содержащие аминокислоты и пептиды, витамины и витаминоподобные вещества, коферменты и микроэлементы, применяются во всех областях медицины и по любому поводу. Такая популярность, очевидно, может объясняться как их эффективностью при лечении разнообразных патологических состояний, так и относительной безвредностью. Это сочетание факторов приводит к тому, что врачу легче назначить тот или иной препарат «на всякий случай», чем разбираться в целесообразности такого назначения. В результате из-за бездумного применения, из-за отсутствия методологической базы страдает эффективность лечения, что, в свою очередь, часто порождает сомнение в его принципиальной результативности. Все это диктует необходимость создания рациональной концепции применения лекарственных средств, относимых к метаболическим [9].
Важная группа таких препаратов представлена так называемыми энерготропными средствами, то есть средствами, усиливающими интенсивность обмена энергии на клеточном уровне. Наибольшее значение в контексте настоящей статьи имеют препараты (в таблице представлены некоторые из них), воздействующие на процессы, происходящие в универсальных клеточных органеллах – митохондриях. Митохондрии выполняют много функций, однако их основная задача – образование молекул аденозинтрифосфата (АТФ) в биохимических циклах клеточного дыхания. Накопленная энергия в последующем используется в других участках клетки.
Нарушения функций митохондрий относятся к важнейшим (часто ранним) этапам повреждения клеток. Эти нарушения ведут к недостаточности энергообеспечения клеток, к нарушению многих других важных обменных процессов, к дальнейшему развитию клеточного повреждения, вплоть до гибели клетки. Для клинициста оценка степени митохондриальной дисфункции имеет существенное значение как для формирования представлений о сути и степени происходящих на тканевом уровне процессов, так и для разработки плана терапевтической коррекции патологического состояния. Степень выраженности патологического процесса в том или ином органе связана со степенью зависимости его тканевых элементов от эффективности аэробного окисления.
Степень выраженности патологического процесса в том или ином органе связана со степенью зависимости его тканевых элементов от эффективности аэробного окисления.
Несмотря на то что в основе митохондриальных заболеваний могут быть сотни первичных биохимических дефектов, основные изученные звенья патогенеза, на которых и основаны современные подходы к коррекции митохондриальной недостаточности, связаны с нарушением реакций окисления пирувата до ацетил-КоА с помощью пируватдегидрогеназного комплекса; окисления ацетил-КоА до углекислого газа и образования восстановленных носителей электронов NADH и FADh3; реокисления восстановленного коэнзима Q ферментами электронно-транспортной цепи внутренней митохондриальной мембраны; транспорта свободных жирных кислот через мембрану митохондрии в виде эфиров карнитина; окислительного дезаминирования аминокислот с последующим поступлением их углеродного скелета в цикл Кребса; перекисного окисления и образования свободных радикалов.
Оценка достоверной эффективности энерготропных препаратов при митохондриальных болезнях сложна по многим причинам [7]. Вариабельность комплексных фенотипов затрудняет сравнение даже двух отдельно взятых больных с одним и тем же заболеванием. Поражение различных органов усложняет сравнительную оценку эффективности результата в целом. Большой проблемой является отсутствие четких критериев оценки динамики заболевания, наиболее выраженные признаки которого – это такие спорадические события, как инсультоподобные эпизоды или судороги.
Все это во многом объясняет тот факт, что, проанализировав в огромной работе 1335 источников о лечении митохондриальных болезней, Джералд Пфеффер (G. Pfeffer) и соавт. [7] смогли отобрать только 12 исследований, строго соответствующих критериям рандомизированного клинического исследования, причем в большинстве случаев они касались воздействия на нервно-мышечные проявления митохондриальных заболеваний с возможностью долговременной оценки таких признаков, как мышечная сила.
Проблема выбора дозы энерготропных препаратов
Сложность проблемы определяется, в частности, двумя факторами: во-первых, бытующим требовательным ожиданием заместительного эффекта при терапии митохондриальных болезней, а во-вторых, недоверием многих клинических биохимиков и фармакологов к возможности легкого введения тех или иных органических молекул внутрь митохондрии. Исходя исключительно из подобных теоретических соображений, ставятся, например, под сомнение как обоснованность применения янтарной кислоты, одного из ключевых метаболитов митохондрий, так и достоверность наблюдаемых позитивных эффектов этого препарата. Однако недавними исследованиями М.Н. Кондрашовой и сотрудников ее школы показано, что терапевтический эффект янтарной кислоты основан не на заместительном принципе, а на сигнальном. Таким образом, чтобы получить эффект, совершенно не нужно заполнять все митохондрии во всех клетках организма янтарной кислотой путем искусственного введения в больших количествах, достаточно назначить микродозы (5–10 мг/кг/сут). В нашей работе, используя новые диагностические приемы с применением транскутанного мониторирования рО2 и рСО2, мы выявили наличие подобного эффекта у L-карнитина. Вполне вероятно, что подобный же принцип может быть применен и к другим лекарственным веществам, используемым в терапии полисистемных нарушений цитоэнергетики.
Кроме того, в настоящий момент недоверие к возможности введения тех или иных молекул в митохондрию значительно поколеблено благодаря открытию большого и сложного комплекса транспортных систем, обслуживающих эти органеллы [10]. Нельзя не упомянуть о работах, показывающих эффективность применения энерготропных препаратов в достаточно высоких дозах. Так, в работе Р. Боулса [11] описана эффективность применения больших доз коэнзима Q10 (10 мг/кг/сут, но не более 200 мг в сутки) и L-карнитина (100 мг/кг/сут, но не более 2 г в сутки) в лечении синдрома циклической рвоты и других состояний, предположительно связанных с митохондриальными дисфункциями, – мигренозной головной боли, миалгии и синдрома множественных локальных болей.
В настоящее время нет единого понимания, какая длительность курса может быть оптимальной при энерготропной терапии. Естественно, во многих случаях (например, при лечении хронических заболеваний) необходимо достаточно длительное лечение, особенно если принимать во внимание вероятность заместительного механизма действия. Однако, исходя из практического опыта многих клиницистов и с учетом рекомендаций патофизиологов, длительное постоянное применение энерготропных препаратов (во всяком случае некоторых) не нужно. Целесообразнее применять схемы с периодическими назначением (1–3 месяца) и отменой (примерно на такой же или несколько больший период). Таким образом, совершенно актуально использование как высокодозовых и длительных, так и низкодозовых кратковременных схем применения энерготропных препаратов. Поскольку выявление сигнальных и заместительных составляющих эффекта для большинства энерготропных препаратов – дело будущего, выбор схемы применения до сих пор зависит от искусства врача.
Коэнзим Q10
Коэнзим Q (коэнзим Q10, убихинон, витамин Q10) – небольшая жирорастворимая молекула, непосредственно участвующая в транспорте электронов по дыхательной цепи митохондрий. Она свободно диффундирует в мембранном бислое и помимо электронов передает ферментному комплексу III также и протоны, которые захватывает из водной среды. Убихинону свойственны витаминоподобные функции. Будучи введенным в организм, он оказывает значительный антиоксидантный эффект, повышает продукцию АТФ и стабилизирует состояние кальциевых каналов.
Коэнзим Q – один из наиболее распространенных и эффективных энерготропных препаратов [12, 13]. В суточных дозах 300–1500 мг он эффективен при дефиците убихинона, дефектах второго и третьего ферментных комплексов дыхательной цепи, клинически выражающихся в синдроме MILS (maternally inherited Leigh syndrome), синдромах MERRF, MELAS и Кернса – Сейра. Его высокая клиническая эффективность в лечении атаксии Фридрейха и других нейродегенеративных заболеваний показана в нескольких работах [14].
Эффект высоких доз коэнзима Q10 (600 мг 2 раза в день перорально в течение двух месяцев) у больных с синдромом MELAS, прогрессирующей наружной офтальмоплегией и некоторыми другими формами митохондриальных болезней был изучен в двойном слепом плацебоконтролируемом исследовании [7, 15]. Было показано, что на фоне увеличения концентраций коэнзима Q10 в крови снижался уровень лактата при кратковременной, но не при долгосрочной нагрузке. Достоверных изменений других биохимических показателей отмечено не было.
Так же как и для многих других энерготропных препаратов, примеры эффективного применения коэнзима Q10 часто можно обнаружить в работах, посвященных терапии состояний, связанных с различными вторичными проявлениями тканевой гипоксии. Так, Д.М. Ароновым и соавт. [16] было проведено рандомизированное проспективное исследование коэнзима Q10 (препарат Кудевита®) в лечении пациентов с ишемической болезнью сердца с сердечной недостаточностью II–III функционального класса. Пациенты основной группы принимали препарат в дозе 150 мг/сут: по 2 капсулы (60 мг) утром и 3 капсулы (90 мг) вечером. Одновременно пациенты основной и контрольной групп получали стандартную терапию, показанную при данном заболевании. В процессе исследования не назначали препараты, влияющие на метаболизм миокарда, кардиопротекторы и антиоксиданты (триметазидин, мельдоний, оксиметилэтилпиридина сукцинат, инозин, аденозинтрифосфат, кокарбоксилаза, витаминные и иные метаболические средства). Длительность наблюдения составила 3 месяца.
В результате было показано, что лечение больных хронической сердечной недостаточностью на фоне ишемической болезни сердца и перенесенного инфаркта миокарда, находящихся на поликлиническом наблюдении, оказало за относительно небольшой отрезок времени существенное влияние на состояние сердечно-сосудистой системы. При этом препарат способствовал снижению диастолического артериального давления, достоверному улучшению физической работоспособности, сократительной функции сердца и гемодинамики по данным ультразвукового исследования (УЗИ), улучшению картины электрокардиограммы (ЭКГ), указывающей на положительные сдвиги в метаболизме миокарда, липидного профиля плазмы крови. Клинически отмечалось уменьшение количества и выраженности приступов боли в груди; уменьшалась частота приема нитроглицерина.
Клинически отмечалось уменьшение количества и выраженности приступов боли в груди; уменьшалась частота приема нитроглицерина.
Препараты, содержащие коэнзим Q10, хорошо знакомы врачам разных специальностей, но до недавнего времени они не могли широко применяться в лечебной практике, так как были представлены в России в виде БАД. Кудевита® в настоящее время является единственным зарегистрированным в России безрецептурным лекарственным препаратом с активным действующим веществом убидекаренон (коэнзим Q10) и, несомненно, поможет более успешно лечить различные заболевания у детей.
L-карнитин
Карнитин – низкомолекулярное соединение, производное аминомасляной кислоты. В тканях млекопитающих присутствует только L-стереоизомер (левокарнитин), именно он биологически эффективен. Карнитин принимает непосредственное участие в катаболизме липидов, обеспечивая перенос длинноцепочечных жирных кислот в виде сложных эфиров (ацилкарнитинов) из цитоплазмы через наружную и внутреннюю митохондриальные мембраны в матрикс митохондрий. Внутри митохондрий транспортированные жирные кислоты подвергаются бета-окислению с образованием ацетил-КоА, который служит субстратом для цикла трикарбоновых кислот Кребса и последующего синтеза АТФ в организме. Наряду с этим окисление жирных кислот – главный путь кетогенеза, а кетоновые тела являются дополнительным энергетическим источником для периферических тканей и головного мозга.
Влияние карнитина на жировой обмен осуществляется также его участием в цитоплазматическом синтезе жирных кислот путем обратного переноса необходимых для этого ацетильных групп митохондриального ацетил-КоА через митохондриальную мембрану в цитоплазму. Помимо перечисленного, карнитин регулирует отношение «ацил-КоА/свободный КоА» в митохондриях. Связывая ацильный радикал, он высвобождает КоА и тем самым активирует интенсивность энергетического метаболизма в тканях. Исключительное значение карнитина становится очевидным в условиях высокого расходования энергетических ресурсов – при заболеваниях, усиленных физических или эмоциональных нагрузках, а также при недостаточном питании. После истощения запасов углеводов липиды становятся главным источником синтеза АТФ в организме.
После истощения запасов углеводов липиды становятся главным источником синтеза АТФ в организме.
Другая важная функция карнитина заключается в его способности образовывать соединения с различными органическими кислотами, являющимися промежуточными продуктами окислительных процессов. Данные вещества, накапливаясь в митохондриях и цитоплазме клеток, оказывают мембранотоксическое действие и ингибируют активность ряда ферментов. Выведение этих токсичных органических соединений из организма происходит через почки в виде ацилкарнитинов.
Левокарнитин высокоэффективен при лечении как первичных форм дефицита карнитина, так и широкого круга заболеваний, связанных со вторичным снижением его содержания в организме. Кроме того, занимая уникальное положение относительно митохондрии и стимулируя приток в нее энергосубстратов, карнитин является универсальным стимулятором тканевого энергообмена, что актуально не только в отношении компенсации энергодефицита, но и в отношении компенсаторной адаптации практически к любым структурно-функциональным дефектам. Данные об эффективности применения препаратов L-карнитина у больных митохондриальными заболеваниями достаточно многочисленны [13, 17], хотя чаще описывают их применение в комплексе с другими энерготропными средствами (см. ниже).
Отдельно следует упомянуть о важности применения L-карнитина при формах его наследственной недостаточности. В качестве примера упомянем работу Е.А. Николаевой и соавт. [18], в которой была показана эффективность применения препарата Элькар® в дозе 800 мг/сут с двухлетнего возраста у больного с первичным системным дефицитом карнитина. Динамическое наблюдение в течение 2 лет показало выраженное улучшение самочувствия и состояния мальчика на фоне терапии Элькаром. Ребенок и его родители не предъявляли жалоб, связанных с лечением препаратом, физическое развитие в возрасте 4 лет было средним (вырос на 19 см), мышечный тонус физиологическим. В результате лечения отмечены положительные изменения со стороны сердца, улучшились биохимические показатели крови.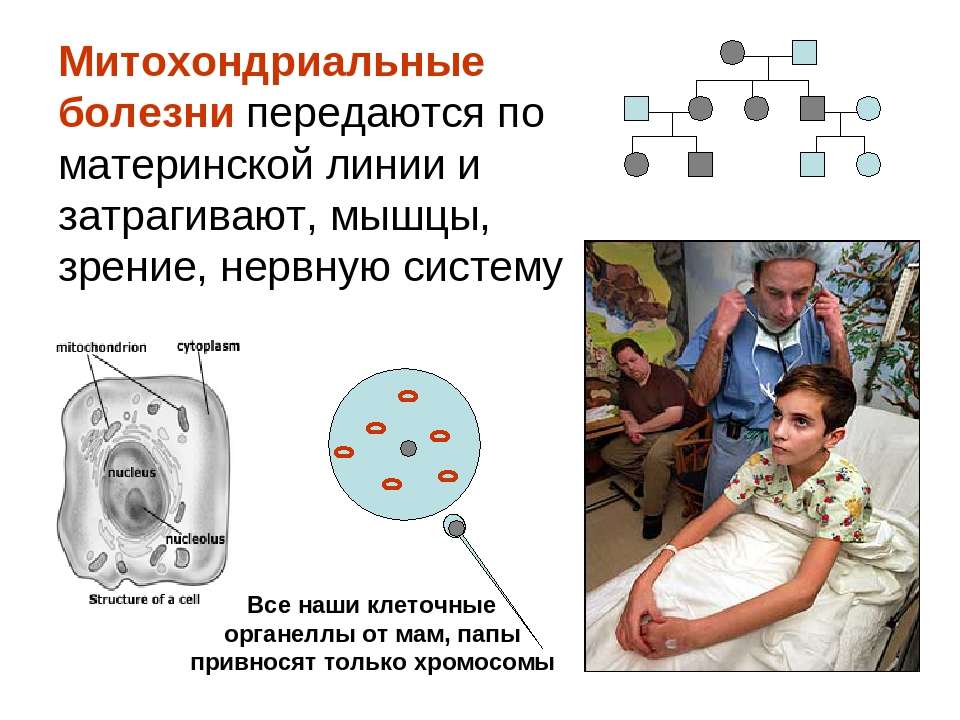
Креатин
Моногидрат креатина представляет собой дополнительный источник энергии. В отличие от нестабильных форм – чистого креатина и фосфата креатина, – моногидрат креатина прекрасно всасывается и с успехом применяется в спортивной медицине. Отмечена его эффективность в лечении различных митохондриальных болезней: синдромов Лея, Кернса – Сейра, MELAS и др. [14, 19–21]. Однако существуют и противоположные данные, не подтверждающие его эффективность при митохондриальных болезнях [12].
В рандомизированном плацебоконтролируемом исследовании [7, 22, 23] эффективности моногидрата креатина, применяемого в течение трех недель (4–10 г в сутки) у больных синдромом MELAS и митохондриальной миопатией, отмечено достоверное увеличение мышечной силы и снижение уровня лактата после нагрузки. В двойных слепых плацебоконтролируемых исследованиях у больных с прогрессирующей наружной офтальмоплегией и митохондриальной миопатией [24] (20 г в сутки в течение месяца) и у больных с хронической прогрессирующей наружной офтальмоплегией и с синдромом Кернса – Сейра [25] (150 мг/кг в течение 6 недель) клиническая эффективность моногидрата креатина не выявлена.
Дихлорацетат
Дихлорацетат – активатор пируватдегидрогеназы – уже значительное время активно изучается в качестве возможного средства исправления митохондриальных функций [12]. В ряде работ показана его эффективность при лечении синдрома MELAS [26, 27], дефицита пируватдегидрогеназного комплекса [28], а также при исправлении митохондриальных функций в опухолевых клетках, ведущем к позитивному эффекту при лечении рака. Некоторые исследователи столь высоко оценивают его лечебный потенциал, что назначают этот препарат несмотря на то, что применение дихлорацетата (в частности в суточных дозах 25 мг/кг) может вызывать развитие периферических полиневропатий [29].
В двойном слепом плацебоконтролируемом исследовании эффективности дихлорацетата у больных с различными митохондриальными заболеваниями [7, 30] (50 мг/кг в сутки в течение недели двумя курсами с интервалом три месяца) было отмечено значительное снижение концентраций лактата, пирувата и аланина в крови (в покое и после нагрузки), а в мозге – значительное снижение соотношения «лактат/креатин» и повышение соотношений «холин/креатин» и «ацетиласпартат/креатин». В других подобных работах [28, 31, 32] была также отмечена нормализация уровня лактата (а в первых двух из этих работ – и пирувата) после приема дихлорацетата.
В других подобных работах [28, 31, 32] была также отмечена нормализация уровня лактата (а в первых двух из этих работ – и пирувата) после приема дихлорацетата.
Янтарная кислота
Сукцинат – один из эффективных медиаторов транспорта электронов, успешно использующийся при острых нарушениях тканевого дыхания. В отношении хронических расстройств целесообразность его применения спорна, однако в научной литературе приводятся описания эффективности сукцината при недостаточности I дыхательного комплекса [33] и при синдроме MELAS (длительная монотерапия суточной дозой 6 г) [34].
Фолиевая кислота
Фолиевая кислота – водорастворимый витамин (В9), необходимый в первую очередь при активной репликации ДНК, то есть делении клеток. Однако описан выраженный положительный эффект этого витамина в суточной дозе 1–2,5 мг/кг при синдроме Кернса – Сейра [33].
L-аргинин
L-аргинин – незаменимая для детей аминокислота, снабжающая азотом систему NO-синтаз. В настоящее время растет число указаний [12] на эффективность применения L-аргинина при синдроме MELAS, в частности, в отношении терапии инсультоподобных эпизодов и сердечно-сосудистых нарушений. Так, например, с использованием позитронно-эмиссионной томографии показано, что применение L-аргинина эффективно при лечении кардиомиопатии при синдроме MELAS [35].
Другие вещества
Наряду с вышеперечисленными, к веществам, несомненно позитивно влияющим на клеточный энергообмен, относят витамин Е (альфа-токоферол), витамин С (аскорбиновая кислота), липоевую кислоту, глутатион, рибофлавин, тиамин и др. Однако в литературе, посвященной митохондриальным болезням, пока не находится четких доказательств их эффективности (по крайней мере, в моноварианте), хотя эти вещества часто используются в комплексных схемах энерготропной терапии.
В настоящее время в различных, в первую очередь экспериментальных, работах активно исследуются новые вещества, представляющие собой потенциально перспективные препараты для лечения митохондриальных заболеваний – антиоксиданты в соединении с трифенилфосфониевым катионом (митохинон, MitoVitE, MitoTEMPOL, MitoPBN, смесь Скулачева), тролокс, SS-пептиды (Szeto-Schiller peptides) [14, 36, 37], ресвератрол [38], препараты, влияющие на сборку дыхательных комплексов [39], оптимизирующие обмен кальция [40], активаторы митохондриального биогенеза [41] и др.
Комплексная энерготропная терапия
Спектр потенциальных патологических нарушений клеточного энергообмена чрезвычайно велик (повреждения различных звеньев цикла Кребса, дыхательной цепи, бета-окисления и др.). Хотя перечень энерготропных препаратов также достаточно широк, далеко не всегда имеется возможность выявить конкретное точечное повреждение митохондрий и точно подобрать подходящий лекарственный препарат. В связи с этим наиболее эффективными в широкой клинической практике могут быть комплексы энерготропных препаратов, обладающих способностью воздействовать сразу на несколько ключевых этапов клеточного энергообмена. При этом на первое место по значимости выдвигаются такие препараты, как L-карнитин, коэнзим Q10, цитохром С и их комплексы с другими вышеперечисленными лекарственными средствами [42–44]. Схемы лекарственной коррекции цитоэнергетической недостаточности у детей активно разрабатываются в настоящее время в Московском НИИ педиатрии и детской хирургии и в Российском национальном исследовательском медицинском университете им. Н.И. Пирогова.
Так, данные Е.А. Николаевой свидетельствуют о том, что при митохондриальных энцефаломиопатиях комплексная энерготропная терапия позволяет добиться существенного клинического эффекта во всех сферах проявления патологического процесса. Результатом лечения является нарастание массы тела, уменьшение выраженности сердечно-сосудистых нарушений, снижение частоты приступов рвоты, судорог, уменьшение выраженности проявлений энцефалопатии и миопатии, снижение утомляемости. Некоторые примеры свидетельствуют о том, что эффективность правильно подобранной энерготропной терапии даже при тяжелых «первичных» митохондриальных синдромах может быть поразительной. В качестве одного из примеров можно привести историю болезни ребенка с синдромом Барта – одним из таких синдромов, клиническая картина которого характеризуется задержкой роста и психомоторного развития, миопатией, кардиомиопатией, нарушениями со стороны крови. Многолетнее лечение комплексом препаратов, включавшим в себя коэнзим Q10, цитохром С, L-карнитин (Элькар) и некоторые другие, привело к тому, что в подростковом возрасте главной проблемой этого мальчика с тяжелым наследственным заболеванием стали попытки избежать постановки на воинский учет.
Многолетнее лечение комплексом препаратов, включавшим в себя коэнзим Q10, цитохром С, L-карнитин (Элькар) и некоторые другие, привело к тому, что в подростковом возрасте главной проблемой этого мальчика с тяжелым наследственным заболеванием стали попытки избежать постановки на воинский учет.
В двойном слепом плацебоконтролируемом исследовании [7, 14, 45] у больных с такими митохондриальными заболеваниями, как синдром Кернса – Сейра, синдром MELAS, хроническая наружная офтальмоплегия, оптическая нейропатия Лебера, митохондриальная нейрогастроинтестинальная энцефаломиопатия, а также с митохондриальными заболеваниями с редкими точковыми мутациями была проведена оценка двухмесячного комплексного применения креатина, коэнзима Q10 и липоевой кислоты. В этой работе выявлено статистически достоверное снижение уровней лактата в плазме крови и 8-изопростана – в моче. У больных с синдромом MELAS отмечено также нарастание массы тела (не за счет жировой ткани). Есть указания [46] на то, что в лечении митохондриальных болезней (как психических, так и соматических нарушений) могут быть эффективны комплексы, содержащие коэнзим Q10 (200–400 мг/сут) и рибофлавин (100–400 мг/сут), в некоторых случаях с добавлением витамина С (1000 мг/сут), витамина Е (400 МЕ/сут), карнитина (2000 мг/сут), креатина (5000 мг/сут) и магнезии (250–500 мг/сут).
При заболеваниях, включающих в свой симптомокомплекс «вторичную» митохондриальную недостаточность, также можно добиться улучшения качества жизни больных. Приведем весьма «эффектный» пример: у низкорослых детей c различными неэндокринными наследственными заболеваниями на фоне лечения энерготропными препаратами – L-карнитином (Элькар), коэнзимом Q10 и другими – удается достичь значительной стимуляции роста – до 6–7 см в год. При некоторых заболеваниях благодаря энерготропной терапии впервые была продемонстрирована возможность относительного успеха в лечении (например, при лечении синдрома Ретта и туберозного склероза отмечено улучшение когнитивных и эмоциональных функций). Существенный позитивный эффект применения энерготропных препаратов наблюдался и в ряде других отделений нашего института: урологическом (при комплексном лечении гидронефроза и гиперактивного мочевого пузыря), ожоговом центре (при реабилитации детей после ожогов), кардиологии (при лечении кардиомиопатий, миокардиодистрофии и нарушений сердечного ритма), пульмонологии (при лечении ряда хронических заболеваний легких) и др.
Существенный позитивный эффект применения энерготропных препаратов наблюдался и в ряде других отделений нашего института: урологическом (при комплексном лечении гидронефроза и гиперактивного мочевого пузыря), ожоговом центре (при реабилитации детей после ожогов), кардиологии (при лечении кардиомиопатий, миокардиодистрофии и нарушений сердечного ритма), пульмонологии (при лечении ряда хронических заболеваний легких) и др.
Применение средств метаболической коррекции позволило оказать существенное влияние на состояние здоровья детей дошкольного возраста с различными вариантами нарушения речевого развития (общее недоразвитие речи, дислалия, задержка психоречевого развития), у детей с соединительнотканной дисплазией и в группе так называемых часто болеющих детей. Эти работы выполнялись нами совместно со специалистами Российского государственного медицинского университета им. Н.И. Пирогова (группа С.О. Ключникова). В лечении этих групп детей были применены комплексы, включающие коэнзим Q10, L-карнитин (Элькар), ряд других энерготропных препаратов. Указанное лечение дети получали, как правило, длительно, в течение 2–3 месяцев, после чего они проходили повторное обследование, позволившее выявить существенную положительную динамику в состоянии здоровья. Отмечались минимизация предъявляемых жалоб, улучшение сна и аппетита, исчезновение или снижение выраженности ряда клинических признаков заболеваний, нормализация лабораторных показателей; возрастала выносливость в отношении физических и интеллектуальных нагрузок.
Заключение
Все вышесказанное свидетельствует о необходимости научно-прикладных разработок, направленных на создание современных принципов энерготропного лечения (по отработке состава энерготропных комплексов, тщательному подбору доз активных веществ, определению оптимальных схем назначения, в том числе с учетом хронобиологических ритмов). Приведенные выше примеры свидетельствуют о необходимости именно комплексного использования таких средств. Однако при каждой нозологической форме должны разрабатываться свои специализированные комплексы, включающие патогенетически наиболее значимые компоненты клеточного энергообмена (например, коэнзим Q10, L-карнитин, цитохром С, янтарная кислота и др.).
Однако при каждой нозологической форме должны разрабатываться свои специализированные комплексы, включающие патогенетически наиболее значимые компоненты клеточного энергообмена (например, коэнзим Q10, L-карнитин, цитохром С, янтарная кислота и др.).
Пронуклеарный перенос ядер может предотвратить митохондриальные заболевания у детей
Пронуклеарный перенос ядер — вспомогательная репродуктивная технология, которую также называют «донорством митохондрий». Его используют в том случае, когда у женщины присутствует высокий риск передачи дефектной митохондриальной ДНК своему ребенку, что может привести к развитию у него тяжелого митохондриального заболевания.
Митохондрии представляют собой небольшие внутриклеточные структуры, которые участвуют в регулировании обмена веществ. Они превращают белки, сахара и жиры в энергию, доступную для использования нашими клетками.
По статистике, 1 из 5 000 детей страдает от тяжелой митохондриальной болезни. Эффективных методов лечения таких болезней пока не существует, поэтому большинство больных детей умирают, не достигнув возраста 5 лет, от дыхательной, сердечной или печеночной недостаточности и других причин, связанных с дефектами в функционировании митохондрий.
Митохондриальные заболевания объединяют около 300 различных генетических нарушений, влияющих на энергетические структуры в клетках организма и, тем самым, оказывающих негативное влияние и на функции органов.
Примерно у 50 % пациентов с митохондриальными заболеваниями причиной этих болезней является проблема одного из 20 000 ядерных генов, которые мы наследуем от каждого из родителей. Это относится и к таким наследственным заболеваниям, как муковисцидоз и талассемия.
У второй половины пациентов митохондриальные заболевания связаны с проблемой в одном из 37 генов, входящих в структуру круговой хромосомы митохондриальной ДНК, и наследуются от матери. Именно в этом случае может помочь донорство митохондрий или пронуклеарный перенос.
Пронуклеарный перенос ядер предусматривает объединение 20 000 уникальных генов ядра клетки матери с таким же количеством генов от отца, но при этом 37 уникальных генов митохондриальной ДНК матери заменяют митохондриями из донорской яйцеклетки. Это позволяет избежать передачи ребенку митохондриальных болезней от матери.
диагностика и подходы к лечению
Список сокращений и условных обозначений
Введение
Глава 1. Органические ацидурии и аминоацидопатии
Глутаровая ацидурия тип I
Метилмалоновая ацидурия
Пропионовая ацидурия
Изовалериановая ацидурия
Множественная карбоксилазная недостаточность с поздним дебютом
Болезнь Канаван
Некетотическая гиперглицинемия
Болезнь с запахом кленового сиропа мочи
Тирозинемия тип 1
Нарушения цикла мочевины
Гомоцистинурия
Список литературы
Глава 2. Митохондриальные заболевания
Митохондриальные заболевания, обусловленные точковыми мутациями мтДНК
Синдром LHON
Синдром NARP
Синдром MERRF
Синдром MELAS
Митохондриальные заболевания, обусловленные крупными перестройками мтДНК
Синдром Кернс-Сейера
Митохондриальные заболевания, связанные с дефектами ядерной ДНК
Мутации генов, кодирующих структурные компоненты КДЦМ или белки, участвующие в их сборке
Синдром Ли
Мутации ядерных генов, приводящие к нарушению стабильности мтДНК
Прогрессирующая полиодистрофия
Альперса в сочетании с циррозом печени
Другие гепатоцеребральные формы, связанные с истощением мтДНК
Множественные делеции мтДНК
СPEO/СPEO+-синдром
Митохондриальная нейрогастроинтестинальная энцефалопатия
Список литературы
Глава 3. Нарушения митохондриального β-окисления жирных кислот
Нарушения транспорта жирных кислот в митохондрии
Недостаточность карнитин-пальмитоилтрансферазы I
Недостаточность карнитин-пальмитоилтрансферазы II
Нарушения β-окисления жирных кислот в митохондриях
Недостаточность очень длинноцепочечной ацил-КоА-дегидрогеназы жирных кислот
Недостаточность митохондриального трифункционального белка
Недостаточность среднецепочечной ацил-КоА-дегидрогеназы жирных кислот
Недостаточность короткоцепочечной ацил-КоА-дегидрогеназы жирных кислот
Глутаровая ацидемия тип II
Список литературы
Глава 4. Лизосомные болезни накопления
Лизосомные болезни накопления
GМ1-ганглиозидоз
GM2-ганглиозидозы
α-Маннозидоз
Муколипидоз, тип II, III
Метахроматическая лейкодистрофия
Болезнь Краббе
Болезнь Фабри
Мукополисахаридозы
Мукополисахаридоз I типа
Мукополисахаридоз II типа
Мукополисахаридоз III типа
Мукополисахаридоз IV типа
Мукополисахаридоз VI типа
Мукополисахаридоз VII типа
Мукополисахаридоз IX типа
Болезнь Гоше
Болезнь Ниманна-Пика тип А, болезнь Ниманна-Пика тип В
Болезнь Ниманна-Пика тип С
Болезнь Помпе
Нейрональные цероидные липофусцинозы
Врожденный нейрональный цероидный липофусциноз
Младенческая форма НЦЛ
Поздние младенческие формы НЦЛ
Классический вариант поздней младенческой формы НЦЛ
Варианты поздней младенческой формы НЦЛ
Юношеские формы нейронального цероидного липофусциноза
Классическая юношеская форма
Взрослая форма нейронального цероидного липофусциноза
Северная эпилепсия
Список литературы
Глава 5. Нарушения гликозилирования
Заболевания, связанные с нарушениями
N-гликозилирования
CDG синдром тип Iа
CDG синдром тип Iс
Заболевания, связанные с нарушениями
О-гликозилирования
Волкер-Варбурга синдром
Мышечно-глазо-мозговой синдром
Множественные нарушения гликозилирования
COG7 недостаточность
Нарушения гликозилирования гликосфинголипидов и глюкозилфосфотидилинозитолов
Недостаточность GM3-синтазы
Список литературы
Глава 6. Пероксисомные заболевания
Нарушения биогенеза пероксисом
Нарушения функции одного пероксисомного белка или фермента
Х-сцепленная
Болезнь Рефсума
Список литературы
Глава 7. Нарушения обмена металлов
Болезнь Менкеса
Гепатолентикулярная дегенерация
Нейродегенеративные заболевания, связанные с нарушением обмена железа в головном мозге
Нейродегенерация, обусловленная недостаточностью пантетенаткиназы
Синдром HARP
Список литературы
Глава 8. Другие наследственные формы лейкоэнцефалопатий/лейкодистрофий
Другие наследственные формы лейкоэнцефалопатий/лейкодистрофий
Болезнь Александера
L-2-гидроксиглутаровая ацидурия
Лейкоэнцефалопатия с макроцефалией с субкортикальными кистами
Лейкоэнцефалопатия с преимущественным поражением ствола мозга, спинного мозга и повышенным лактатом при МР-спектроскопии
Лейкоэнцефалопатия с «исчезающим» белым веществом
Список сокращений и условных обозначений
EPILEPSY IN CHILDREN WITH MYTOCHONDRIAL DISEASES: DIAGNOSTICS AND TREATMENT FEATURES | Zavadenko
1. Мазунин И.О., Володько Н.В., Стариковская Е.Б., Сукерник Р.И. Митохондриальный геном и митохондриальные заболевания человека. Молекулярная биология. 2010; 44 (5): 755-772.
2. Михайлова С.В., Захарова Е.Ю., Петрухин А.С. Нейрометаболические заболевания у детей и подростков: диагностика и подходы к лечению. М.: Литтерра, 2011, 341 с.
3. Мутовин Г.Р. Клиническая генетика. М.: ГЭОТАР-Медиа, 2010, 832 с.
4. Сухоруков В.С. Очерки митохондриальной патологии. М.: Медпрактика-М, 2011, 287 с.
5. Bindoff LA. Mitochondrial function and pathology in status epilepticus. Epilepsia 2011; 52 (suppl. 8): 6-7.
6. Canafoglia L, Franceschetti S, Antozzi C, Carrara F, Farina L, Granata T, Lamantea E, Savoiardo M, Uziel G, Villani F, Zeviani M, Avanzini G. Epileptic phenotypes associated with mitochondrial disorders. Neurology 2001; 56: 1340-1346.
7. Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann. Neurol. 2001; 49: 377-383.
The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann. Neurol. 2001; 49: 377-383.
8. El Sabbagh S, Lebre A-S, Bahi-Buisson N, Delonlay P, Soufflet C, Boddaert N, Rio M, Rotig A, Dulac O, Munnich A, Desguerre I. Epileptic phenotypes in children with respiratory chain disorders. Epilepsia 2010; 51: 1225-1235.
9. Finsterer J. Mitochondriopathies. Eur. J. Neurol. 2004; 11: 163-186.
10. Lagrue E, Chalon S, Bodard S, Saliba E, Gressens P, Castelnau P. Lamotrigine is neuroprotective in the energy deficiency model of MPTP intoxicated mice. Pediatr. Res. 2007; 62: 14-19.
11. Lee Y., Kang H., Lee J., Km S., Kim E., Lee S., Slama A., Kim H. Mitochondrial respiratory chain defects: underlyingetiology in various epileptic conditions. Epilepsia. 2008; 49: 685-690.
12. Lheureux P.E., Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. ClinToxicol (Phila). 2009; 47: 101-11.
13. Mancuso M., Galli P., Pizzanelli C., Filosto M., Siciliano G., Murri L. Antimyoclonic effect of levetiracetam in MERRF syndrome. J. Neurol Sci. 2006; 243: 97-99.
14. Schaefer A.M., McFarland R., Hart Y., Turnbull D.M. Newcastle Mitochondrial Disease Guidelines. Newcastle Mitochondrial Centre, NHS Specialised Services for Rare Mitochondrial Disorders of Adults and Children. 2010; 17 p.
15. Siekevitz P. Powerhouse of the cell. Scientific American 1957; 1: 131-140.
Siekevitz P. Powerhouse of the cell. Scientific American 1957; 1: 131-140.
16. Zammit V.A., Ramsay R.R., Bonomini M., Arduini A. Carnitine, mitochondrial function and therapy. Adv. Drug. Deliv. Rev. 2009; 61: 1353-1362.
Откуда у девочки Аланы три родителя
- Шарлотт Притчард
- Би-би-си
Алана Сааринен любит играть в гольф и на фортепиано, слушать музыку и общаться с друзьями. В этом отношении она мало отличается от других подростков. Но кое в чем Алана отличается от большинства людей: каждая клетка ее тела содержит ДНК трех человек. Таких людей в мире совсем немного.
«Многие говорят, что черты лица у меня от мамы, глаза от папы… Я унаследовала от них некоторые признаки, и характер тоже», — говорит Алана.
«Но у меня есть ДНК от еще одной женщины. Ее я третьим родителем не считаю, у меня просто митохондрии от нее», — замечает девочка.
Митохондрии часто называют энергетическими станциями клетки. Они вырабатывают энергию, необходимую для деятельности каждой клетки, и тем самым поддерживают функционирование организма. Но еще в них содержится небольшое количество ДНК.
В мире живет всего лишь от 30 до 50 человек, рожденных из яйцеклеток, в которые были пересажены митохондрии, а вместе с ними и ДНК, от третьего лица. Алана – одна из них. Она была зачата в США в рамках эксперимента по лечению бесплодия. Метод, применявшийся в ходе этого эксперимента, впоследствии был запрещен.
Но, возможно, в скором времени людей с тремя биологическими родителями, как у Аланы, станет больше, поскольку в Великобритании рассматривается идея легализации нового, похожего метода, предусматривающего использование донорских митохондрий для лечения опасных генетических заболеваний.
Новый метод называется митохондриальной заменой. Если парламент одобрит его применение, Великобритания станет единственной страной в мире, допускающей рождение детей с тремя родителями.
Цитоплазматический перенос
Подпись к фото,
Шарон Сааринен (на фото слева) не нарадуется на свою дочь
Метод лечения бесплодия, благодаря которому Алана появилась на свет, называется цитоплазматическим переносом. Ее мать, Шарон Сааринен, 10 лет пыталась зачать. Перепробовала все существовавшие тогда методы искусственного оплодотворения, — но все безуспешно.
«Я чувствовала себе никчемной. Я испытывала чувство вины перед мужем за то, что не могла дать ему ребенка. Когда хочешь родить биологического ребенка, но не можешь, возникает чувство отчаяния. Невозможно заснуть, это с тобой постоянно, занимает все твои мысли», — вспоминает женщина.
В конце 1990-х годов клинический эмбриолог доктор Жак Коэн и его команда из Института Сент-Барнабус в Нью-Джерси (США) предложили технологию цитоплазматического переноса.
«Мы чувствовали, что есть шанс, что какой-то элемент, какая-то структура в цитоплазме клетки функционирует не оптимально. И одним из главных кандидатов […] были структуры митохондрий», — рассказывает Коэн.
Коэн пересадил содержавшую митохондрии цитоплазму из клеток женщины-донора в яйцеклетку Шарон Сааринен. После этого он оплодотворил ее яйцеклетку спермой ее мужа. В оплодотворенную яйцеклетку попали и донорские митохондрии, а с ними и ДНК.
В клинике Коэна методом цитоплазматического переноса было зачато 17 младенцев, несущих в себе, таким образом, ДНК трех человек.
Опасность аномалий
Применение этого метода вызывало определенные опасения. Всего забеременело 12 женщин, но у одной из них беременность закончилась выкидышем на раннем сроке.
Коэн и его коллеги полагают, что выкидыш был обусловлен отсутствием X-хромосомы и что в принципе один случай из 12 – это ожидаемый процент.
«Там еще было зачатие близнецов, и один из них родился совершенно нормальным, а у второго отсутствовала X-хромосома. То есть две [аномалии] на всю эту небольшую группу зародышей, зачатых в результате применения этой процедуры. Но и это нас беспокоило, и мы должным образом отразили этот факт в литературе и сообщили комитету по этике, надзирающему за нашей деятельностью», — говорит ученый.
Все прочие дети на момент рождения представлялись здоровыми, но через год-два у одного из них выявились «ранние признаки комплексных нарушений развития, включавших в себя широкий спектр когнитивных расстройств, в том числе аутизм», отметил Коэн.
По его словам, трудно сказать, были ли эти отклонения от нормы случайностью или же закономерным следствием цитоплазматического переноса.
Метод переняли другие клиники, и, по оценке Коэна, таким путем в мире родилось от 30 до 50 детей с ДНК трех человек.
Однако в 2002 году американское Управление по безопасности пищевых продуктов и лекарственных средств потребовало прекратить применение метода цитоплазматического переноса, до тех пор пока не будет доказана его этичность и безопасность. Все клиники выполнили требование надзорного органа.
«Была реакция ученых, специалистов по этике, общественности. В основном реакция была позитивная, но была и критика. По-моему это нормально. Каждый раз, когда в медицине проводится эксперимент, возникает реакция: а каковы риски?» – объясняет Коэн.
Основной момент, на который указывали критики, заключался в том, что эксперимент воздействовал и на структуру уже оплодотворенной клетки, тогда как прежние методы затрагивали лишь структуру сперматозоидов и яйцеклеток. Алана может передать свой необычный генетический код своим детям, а те, в свою очередь, следующим поколениям. Последствия этого пока не изучены.
Новый метод
Из-за недостаточности финансирования не было возможности отследить дальнейшую судьбу детей, родившихся на свет, как Алана, в результате цитоплазматического переноса, говорит Коэн.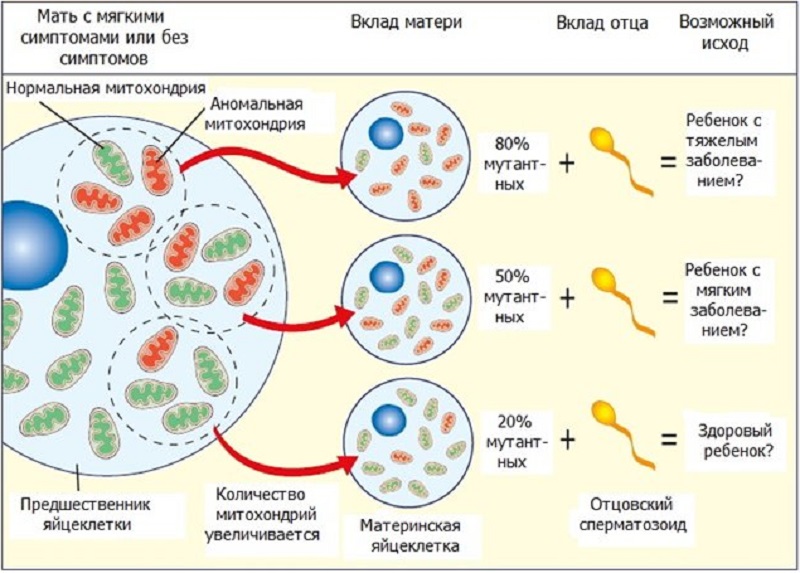 Однако недавно Институт Сент-Барнабус все же нашел необходимые средства.
Однако недавно Институт Сент-Барнабус все же нашел необходимые средства.
Митохондрии наследуются только от матери, поэтому только девочки могут передать свой необычный генетический код.
Шарон Сааринен говорит, что ее дочь – совершенно типичный подросток и пышет здоровьем.
«Она всегда была здоровой. Никогда не болела ничем серьезнее простуды или гриппа. Вообще никаких проблем со здоровьем», — замечает Сааринен.
Здоровье детей, родившихся путем цитоплазматического переноса, вызывает пристальное внимание потому, что митохондриальная замена, которую предлагают легализовать в Великобритании, предназначена для совсем другой цели, нежели лечение бесплодия.
Этот метод рассчитан на женщин с болезнями, вызванными дефектами митохондриальной ДНК. Митохондриальная замена призвана пересадить им здоровую ДНК, чтобы они не могли передать наследственные заболевания потомству.
Потенциально возможно два метода митохондриальной замены:
- Берутся яйцеклетки матери с дефектными митохондриями и донора со здоровыми. Из обеих клеток удаляется ядро, содержащее основную массу генетического материала. Ядро донорской клетки уничтожается. Затем материнское ядро пересаживается в здоровую донорскую яйцеклетку, и та, в свою очередь, оплодотворяется спермой отца.
- Отцовской спермой оплодотворяются обе яйцеклетки – и материнская, и донорская, так что получается два эмбриона. После этого из обеих оплодотворенных яйцеклеток извлекается формирующееся ядро. Ядро, производное от донорской клетки, уничтожается, а на его место пересаживается материнское ядро.
Митохондриальные болезни
«Митохондриальные заболевания обычно поражают ткани и органы, поглощающие большое количество энергии», — говорит профессор Даг Турнбулл из Университета Ньюкасла. Он уже не один десяток лет лечит людей с такими генетическими отклонениями и участвовал в создании технологий лечения от этих болезней, – а они, по его словам, причиняют людям много страданий.
«Заболевания затрагивают сердце, мозг, иногда скелетные мышцы. У людей появляются серьезнейшие пороки сердца, приводящие, в конечном счете, к его остановке, крайняя слабость, при которой люди не могут жить без дыхательных аппаратов или передвигаться без инвалидной коляски. Если болезнь поражает мозг, то отмечается эпилепсия, инсульты, а позже острые формы приобретенного слабоумия», — говорит профессор.
По его оценке, митохондриальными болезнями в Великобритании страдает один человек из 3-5 тысяч. «Мы лечим симптомы. Мы можем улучшить качество и продолжительность жизни таких пациентов, но излечить их мы не можем», — добавляет Турнбулл.
При этом по его словам, в митохондрии содержится всего 13 «важных генов». Для сравнения, в ядре клетки – 23 тыс. генов, определяющих важнейшие физические и психические свойства человека.
«Мы не пытаемся искусственно сделать человека сильнее, или чтобы у него были белокурые волосы. Мы лишь пытаемся предотвратить серьезную болезнь, и это единственное обоснование», — говорит Турнбулл.
Автор фото, Charlotte Pritchard
Шарон Бернарди живет в Сазерленде на севере Англии. Стены ее гостиной увешаны фотографиями ее детей. Все они умерли.
«Мои дети похоронены на трех разных кладбищах, — говорит она. – На это не рассчитываешь, когда пытаешься завести семью. У меня остались чудные фото и воспоминания, но больше ничего».
Почти все дети Бернарди умерли спустя считанные часы после рождения. Врачи не знали, в чем причина. Поэтому Бернарди не оставляла попытки завести здорового ребенка.
Четвертый ее ребенок, Эдвард, сразу не умер. Напротив, он благополучно, как казалось, дожил до четырех с половиной лет. Тут-то ему и поставили диагноз – синдром Лея. Это редкая митохондриальная болезнь. С годами ему становилось все хуже.
«С 20 лет Эдварду стало труднее передвигаться. У него появились новые симптомы – спазмы. Он кричал… по четыре, пять, шесть часов подряд. У него сводило мускулы, руки, лицо. Это было похоже на дистонические спазмы – они очень болезненные. Он по восемь часов кричал от боли. Лицо корчилось, руки немели и не слушались. Смотреть на это было тяжело», — рассказывает Бернарди.
Он кричал… по четыре, пять, шесть часов подряд. У него сводило мускулы, руки, лицо. Это было похоже на дистонические спазмы – они очень болезненные. Он по восемь часов кричал от боли. Лицо корчилось, руки немели и не слушались. Смотреть на это было тяжело», — рассказывает Бернарди.
Три года назад, прожив всего 21 год, Эдвард скончался.
«Чтобы Эдвард умер не напрасно»
Автор фото, BBC World Service
«Вся моя жизнь была посвящена Эдварду. Даже сейчас, когда я засыпаю, я иногда вдруг просыпаюсь от мысли: «Что-то тихо». И только потом вспоминаю, «Брось, Эдварда больше нет», — говорит Шарон Бернарди.
«Я бы без колебаний согласилась на [митохондриальную замену]. Надеюсь, что этот новый вариант кому-то поможет. И что к нему отнесутся серьезно и разрешат», — говорит она.
«Я не хочу, чтобы получилось так, что мой сын умер ни за что. Я хочу, чтобы его смерть сыграла положительную роль. Он лишился жизни в 21 год. Мы хотим, чтобы такого больше не было. Люди должны понять, что эта болезнь калечит всю жизнь. Мы хотим, чтобы она не передавалась по наследству, и тем самым избавить от нее будущие семьи», — подчеркивает Бернарди.
Однако не все настроены столь однозначно. Некоторые считают, что это первый шаг на скользком пути к созданию генетически модифицированных людей.
Фиона Брюс, лидер британской межфракционной парламентской группы за жизнь, говорит, что в случае одобрения новый метод позволит изменять гены на уровне оплодотворенной клетки, а это «определяется в Хартии ЕС об основных правах как по сути евгеника».
«Мы разрешим эту технологию, а для чего она будет использоваться в будущем, — кто знает? Мы открываем ящик Пандоры», — указывает политик.
Решение относительно нового метода предстоит принять британскому Управлению по оплодотворению человека и эмбриологии. Оно заказало три независимых анализа для изучения его безопасности.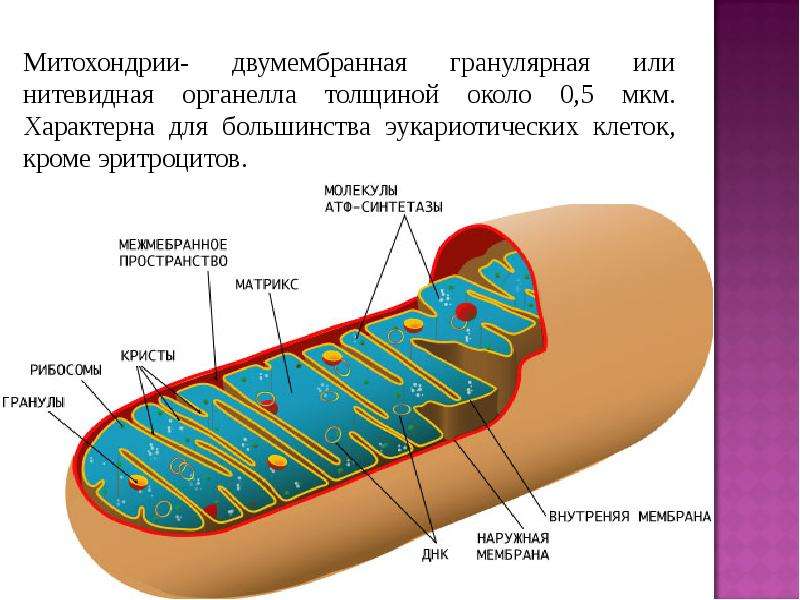 Исследования пришли к выводу, что митохондриальную замену «нет оснований считать небезопасной».
Исследования пришли к выводу, что митохондриальную замену «нет оснований считать небезопасной».
Это значит, что «после некоторых дополнительных экспериментов будет разумно ввести этот метод в клиническую практику при наличии всех необходимых обстоятельств», говорит Питер Брод, почетный профессор акушерства и гинекологии в лондонском Кингс-Колледже. Он участвовал во всех трех группах, анализировавших безопасность митохондриальной замены.
Элемент риска
«В любом переходе от научных исследований к клинической практике есть элемент риска – на него приходится идти», — отмечает Брод.
Он указывает, что когда внедряли экстракорпоральное оплодотворение (ЭКО), высказывались все те же самые опасения. Великобритания уже десятки лет находится на переднем крае вспомогательных репродуктивных технологий, именно здесь еще в 1978 году родилась первая девочка, «зачатая в пробирке», — Луиз Браун.
«И тогда тоже в газетах писали, что мы «играем в бога» и создаем «генетически модифицированных людей», — вспоминает Брод.
«В истории вспомогательных репродуктивных технологий мне известно очень мало случаев, когда новые методы приходилось запрещать из-за опасности. Напротив, насколько я знаю, все идет как по маслу», — указывает он.
Митохондриальная замена, по словам ученого, подвергнется такому же тщательному анализу, как и другие, теперь уже широко применяемые методы, в том числе ЭКО.
«Изначально все техники внедрялись сразу после экспериментов на мышах, кроликах и других лабораторных животных. А здесь все испытывалось на приматах, обезьянах-макаках, и все это было в высшей степени полезно и обнадеживающе. Вот поэтому мы и пришли к выводу, что нет оснований считать митохондриальную замену небезопасной», — отмечает Брод.
Эксперименты на макаках проводились в штате Орегон в США. Родившиеся в ходе эксперимента обезьяны прожили уже пять лет и вроде бы вполне здоровы.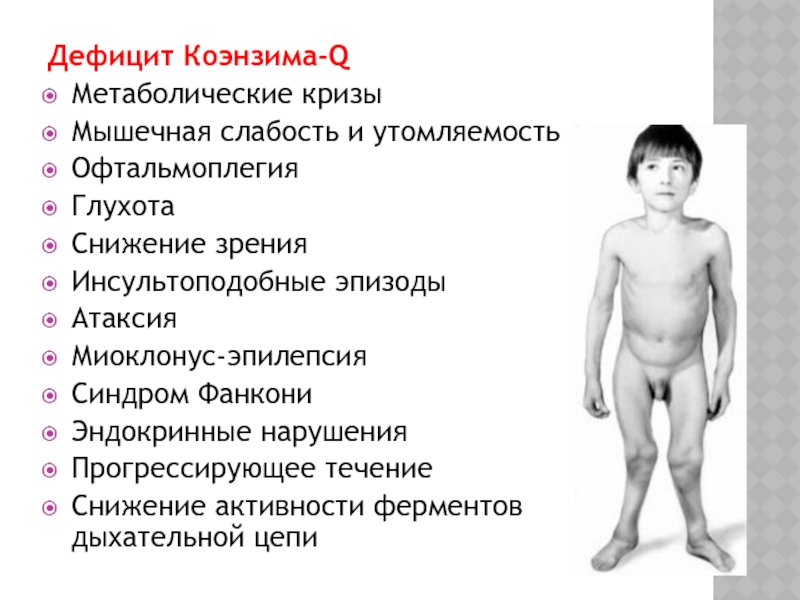
Брод отмечает, что само по себе присутствие ДНК третьего лица в организме не несет в себе ничего нового.
«Представьте себе пересадку костного мозга, допустим, вам не повезло заболеть лейкемией, ваш костный мозг убили облучением вместе с раковой опухолью, а на его место пересадили донорский – например от меня. По сути с этого времени в вашем организме будет содержаться моя ДНК. Но это не будет означать, что вы мой родственник. Вы будете мне благодарны, а ведь у вас в теле появится ДНК третьего лица», — указывает Брод.
Голос скептиков
Критики указывают на то, что в случае митохондриальной замены эта чужеродная ДНК будет передаваться будущим поколениям.
Доктор Тед Морроу из Университета Сассекса и его коллеги проводили эксперименты по митохондриальной замене на животных и в ряде публикаций указывали на определенные риски.
«У мышей, — говорит он, — отмечались изменения в когнитивных способностях – способностях учиться и задействовать мозг. У мух-пестрокрылок изменялась фертильность самцов, менялись процессы старения, в разных экспериментах затрагивался широкий спектр признаков».
Выводы Морроу обсуждались в исследованиях, заказанных Управлением по оплодотворению человека и эмбриологии, но их посчитали не относящимися к людям, поскольку эксперименты проводились на животных, подвергавшихся инбридингу.
Однако Морроу настаивает на актуальности своих исследований и считает, что от них не следовало так легко отмахиваться.
Его данные подхватили противники митохондриальной замены, такие как Фиона Брюс, потребовавшая провести специальные дебаты по этому вопросу в палате общин. «Сам по себе этот метод может привести к тому, что ребенок унаследует неизученные медицинские осложнения», — указывает она.
Морроу находит странным то, что в прессе его изображали союзником групп, выступающих против репродуктивной медицины в целом и абортов в частности. «Я ученый, и я не с теми, кто «за жизнь». Я просто считаю, что здесь и правда есть опасения по поводу безопасности», — говорит он.
«Я ученый, и я не с теми, кто «за жизнь». Я просто считаю, что здесь и правда есть опасения по поводу безопасности», — говорит он.
«Я все сделала правильно»
Подпись к фото,
Алана Сааринен не придает большого значения донору митохондрий в своей жизни
Алана и Шарон Сааринен с интересом следят за британскими дебатами из США.
«Я бы хотела встретиться с ней, с женщиной-донором, чтобы сказать, как я ей благодарна за то, что она для нас сделала. Как отблагодарить человека, который дал тебе жизнь? Это же невозможно», — говорит Шарон.
Алана соглашается с матерью, но осторожно: «Да, я думаю, что было бы хорошо сказать ей спасибо. Но я не хотела бы завязывать с ней какие-то отношения или связи. ДНК от нее у меня совсем немного».
Шарон, однако, не унимается: «Я знаю, что у моей дочери митохондрии другого человека, но посмотрите, какой из нее замечательный человек вырос, и здоровая! А что она передаст гены своим детям, меня нисколько не беспокоит. Я знаю, что все сделала правильно. Каждый день у меня перед глазами живое доказательство того, что все кончилось хорошо».
Митохондриальные заболевания, комплексная диагностика: митохондриальная ДНК, ч. м. (Mitochondrial Diseases, multiplex mutations detection assay)
Исследуемый материал
Цельная кровь (с ЭДТА)
Метод определения
Полимеразная цепная реакция (ПЦР).
Митохондриальные заболевания клинически представляют собой гетерогенную группу состояний, развивающихся в связи с нарушением работы ферментов дыхательной цепи митохондрий. Нарушения могут быть связаны как с мутациями, появляющимися в геноме человека, так и с мутациями в митохондриальной ДНК (мтДНК). Наиболее часто наблюдаются повреждения мтДНК, которые характеризуются протяженными делециями и дупликациями, а также точечными мутациями (чаще всего 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C).
Митохондриальные заболевания могут манифестировать в любом возрасте. При некоторых митохондриальных заболеваниях поражается только один орган (наследственная оптическая нейропатия Лебера), однако в большинстве данная группа состояний имеет системные проявления, чаще всего связанные с поражением нервной и мышечной системы. Довольно часто клинические проявления митохондриальных заболеваний могут подпадать под специфические синдромы: MELAS-синдром (митохондриальная миопатия, энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды), MERRF-синдром (миоклоническая эпилепсия, ассоциированная с порванными красными волокнами), прогрессирующая наружная офтальмоплегия, синдром Кернса-Сейра, синдром Лея, нейрогенная слабость с атаксией и пигментным ретинитом. Однако довольно много клинических случаев митохондриальных заболеваний не подпадают ни под одну из перечисленных категорий, имея перемежающийся спектр проявлений и симптомов, что возможно объясняется гетероплазмией мутаций в мтДНК (вариация количества «мутантных» митохондрий в одной клетке).
Частыми клиническими проявлениями митохондриальных заболеваний являются блефароптоз, офтальмоплегия, проксимальная миопатия, кардиомиопатия, нейросенсорная тугоухость, атрофия глазного нерва, пигментная ретинопатия, энцефалопатия, судороги, деменция, мигрень, инсультоподобные эпизоды, атаксия, спастичность.
Мутации в мтДНК могут передаваться по материнской линии. При наличии мутаций в мтДНК мужчины риск передачи аберрации детям отсутствует. Делеции в мтДНК чаще всего возникают de novo (единственный случай в семье), тогда как точечные аберрации и дупликации часто передаются по материнской линии.
Литература
- Arpa J. et al. Prevalence and progression of mitochondrial diseases: a study of 50 patients. Muscle Nerve. 2003;28:690-695.
- Chinnery P.F. et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592-596.

Терапия зла
Некоторые генетические болезни мы могли бы лечить еще 20 лет назад — методом пересадки митохондрий. Но реальные попытки его применить закончились обвинениями ученых в евгенике, скандалами и запретами, а митохондриальные болезни так и остались неизлечимыми. С тех пор биотехнология шагнула вперед, у нас появились системы редактирования генома и первые пациенты, чьи гены удалось переписать. Время совершить очередной подход к митохондриальным генам. Сможем ли мы на этот раз обойтись без скандалов?
Первые шаги
Когда в августе 1996 года врачи из клиники в Нью-Джерси ввели сперматозоиды мистера Отта в 14 яйцеклеток миссис Отт, никто еще не знал, какая из них превратится в маленькую Эмму и чем закончится эта история для пациентов с митохондриальными болезнями. Тогда супружеская пара Оттов готова была на любые риски после 6,5 лет тщетных попыток зачать ребенка, а доктор Жак Коэн надеялся на успех своей новой методики. Суть ее была проста: в процессе искусственного оплодотворения в яйцеклетку матери врачи ввели не только сперматозоид отца, но и десятую часть цитоплазмы из яйцеклетки молодой женщины-донора.
Из 14 яйцеклеток, оплодотворенных таким образом, шесть начали развиваться нормально, четыре подсадили в организм матери, одна прижилась, выросла в Эмму Отт и родилась в срок без осложнений. Коэн с коллегами отчитались в журнале The Lancet о том, что им успешно удалось восстановить фертильность 39-летней женщины, предыдущие зародыши которой развивались неправильно. Нью-Йоркские газеты вовсю рекламировали их успехи. Десятки бесплодных супружеских пар обращались в клинику за помощью, и за следующие четыре года на свет появились еще 16 подтверждений того, что методика работает.
Нью-Йоркские газеты вовсю рекламировали их успехи. Десятки бесплодных супружеских пар обращались в клинику за помощью, и за следующие четыре года на свет появились еще 16 подтверждений того, что методика работает.
А потом грянул гром.
Коэн и коллеги продолжали совершенствовать свою методику и следить за результатами. В 2000 году они обнаружили, что в разных зародышевых тканях и клетках новорожденных, которые появились на свет в результате пересадки цитоплазмы, остались следы донорских генов.
Эмбрионы на третий день после оплодотворения. (a) яйцеклетка матери + сперматозоид отца; (b) яйцеклетка матери + сперматозоид донора; (c) яйцеклетка донора + сперматозоид отца, (d) яйцеклетка матери + сперматозоид отца и инъекция ооплазмы донора
Carol Brenner et al. / Fertility and Sterility, 2000
Возможно, это наблюдение и прошло бы незаметно, если бы в 2001 году они не опубликовали еще один короткий отчет, посвященный долгосрочным наблюдениям за детьми. На этот раз они нашли следы донорских генов в крови и слизистой щеки у двух годовалых младенцев и честно объявили: «это первый случай наследуемой генетической модификации» — чем и загубили все дело.
Фраза продолжалась словами «… которая привела к рождению нормальных здоровых детей», но это уже никого не волновало.
СМИ бросились обсуждать «первых в мире ГМ-детей» и говорить о возвращении евгеники. FDA, американский аналог Росздравнадзора, потребовало от репродуктивных клиник считать использование донорских яйцеклеток экспериментальной процедурой и получать на них специальное разрешение. Возведя «бумажную стену», бюрократия обуздала технологию и фактически похоронила спорный метод.
Чужой внутри
Сам же Коэн не ставил своей целью создание генетически модифицированных людей и даже не признавал свой метод модификацией — все гены ребенка остались на месте и никак не изменились. Он просто считал, что причина бесплодия кроется в постаревших яйцеклетках женщин и искал способ их омолодить. Более того, врачи из его команды специально следили, чтобы в микрокапилляр (с помощью которого в яйцеклетку вводили сперматозоид и донорскую цитоплазму) не попали чужие хромосомы — от донора им нужна была только цитоплазма, и ее забирали с той стороны яйцеклетки, где не было генетического материала. С этой частью процедуры они справились успешно: в крови детей никаких чужеродных ядерных генов впоследствии не обнаружили.
Он просто считал, что причина бесплодия кроется в постаревших яйцеклетках женщин и искал способ их омолодить. Более того, врачи из его команды специально следили, чтобы в микрокапилляр (с помощью которого в яйцеклетку вводили сперматозоид и донорскую цитоплазму) не попали чужие хромосомы — от донора им нужна была только цитоплазма, и ее забирали с той стороны яйцеклетки, где не было генетического материала. С этой частью процедуры они справились успешно: в крови детей никаких чужеродных ядерных генов впоследствии не обнаружили.
Инъекция сперматозоида в яйцеклетку
СС0
Однако вместе с донорской цитоплазмой в зародыш могли попасть и другие части яйцеклетки, в том числе, митохондрии. Сами по себе они могут быть даже полезны: добавочные митохондрии могут снабдить развивающуюся яйцеклетку дополнительной энергией.
У митохондрий внутри есть собственный геном. Именно его и нашел Коэн в клетках детей, что побудило его использовать столь напугавшее приличную общественность словосочетание «генетическая модификация».
Гетероплазмия — соседство нескольких типов митохондрий в одной клетке — сама по себе не влияет на внутриклеточную жизнь. Более того, она естественным образом появляется в стареющих клетках человека, по мере того как митохондрии накапливают мутации. Поэтому нет никаких причин думать, что чужая митохондриальная ДНК могла повлиять на судьбу и развитие детей. В 2016 году Коэн и коллеги отчитались о здоровье уже выросших «экспериментов»: никаких серьезных аномалий развития, никаких тяжелых болезней, хорошие оценки в школе.
(a) Яйцеклетки через 10 минут после инъекции донорской ооплазмы (красная) (b) Трипронуклеарные зиготы через 24 часа после инъекции донорской ооплазмы. По мнению ученых, красные точки это именно митохондрии
По мнению ученых, красные точки это именно митохондрии
Jason A. Barritt et al. / Human Reproduction, 2001
Но научное сообщество волновало не только здоровье детей. Гораздо более важным аргументом стал тот факт, что часть этих детей — в том числе и «первенец» Коэна Эмма Отт — девочки, а значит, могут передать свой необычный митохондриальный состав по наследству, положив начало клану «неестественно» гетероплазмичных людей.
С тех пор появились свидетельства того, что гетероплазмия в клеточных культурах бывает обратимой, и пришлые митохондрии на чужбине постепенно вымирают. Но многие участники исследований Коэна отказались проверять кровь своих взрослых детей на гетероплазмию, и мы едва ли теперь узнаем, насколько состоятельны были опасения FDA. Запрет регулятора остается в силе по сей день, и ученым пришлось искать обходные пути к лечению бесплодия.
Вторая мать
Коэн так и не смог сказать наверняка, какая именно часть донорской цитоплазмы если не омолодила яйцеклетки, то хотя бы помогла женщинам забеременеть. Это могли быть не только органеллы, но и какие-нибудь отдельные молекулы из молодой цитоплазмы, например, белки или информационные РНК. Тем не менее, работа ученого создала важный прецедент: для создания ребенка можно использовать донорский материал третьего человека. И как только его эксперименты заглохли под пристальным взглядом FDA, дальнейший прогресс переехал в Китай.
Вскоре после того, как FDA изменили правила игры, конкуренты Коэна перенесли свои эксперименты из Нью-Йорка в Гуанчжоу, где никаких запретов еще не существовало. Там молодому эмбриологу Джону Чжану пришло в голову сделать все наоборот: если можно пересадить участок цитоплазмы из молодой яйцеклетки в старую, то почему бы не попробовать сделать наоборот — пересадить ядро старой яйцеклетки в молодую? Технологию переноса ядер (позже ее назвали переносом пронуклеусов) он опробовал в 2003-м: оплодотворил старую (материнскую) и молодую (донорскую) яйцеклетки, затем из второй удалил ядро и пересадил туда ядро первой.
(a) Перенос веретена (мексиканский эксперимент Чжана) (b) Перенос пронуклеусов (китайский эксперимент Чжана))
Steve Connor / Nature, 2017
Насколько эксперимент оказался успешным, сказать сложно. В культуре начали развиваться сразу пять эмбрионов, которые и перенесли пациентке. Из них прижились сразу три. Ученые решили, что это опасно, и вызвали аборт одного из зародышей, а остальные два позже погибли сами. Поэтому Чжан, в отличие от Коэна, не смог доказать, что его методика безопасна. Эксперименты снова запретили — на этот раз уже китайские регуляторные органы, мотивируя это подозрительной близостью исследований к попыткам клонировать человека (а вот оно в Китае запрещено).
Но история, естественно, на этом не закончилась: эту спорную терапию бесплодия (перенос пронуклеусов) продолжают использовать и сейчас. В 2016 году ее начали применять в Украине, в 2019 первый такой ребенок появился в Греции.
Смена курса
Те же, кто не верил в то, что митохондрии могут «омолодить» яйцеклетку, наметили еще один потенциальный выхлоп из этого метода. Перенос пронуклеусов мог бы стать избавлением от мутаций в митохондриальных генах. Довольно часто такие мутации делают своих носителей инвалидами в раннем возрасте: поскольку митохондрии поставляют в клетки энергию, страдают чаще всего главные ее потребители — мышцы и нервы. Носительница таких мутаций не может зачать здоровых детей естественным путем, так как с митохондриями отец помочь никак не может: их ребенок наследует строго от матери.
Таким образом, перенос пронуклеусов можно было использовать как терапию митохондриальных болезней. На это обратили внимание сразу несколько исследовательских групп. Американский биолог русского происхождения Шухрат Миталипов, известный как пионер редактирования генома человека, еще в 2013 году основал компанию Mitogenome therapeutics и начал проверять методику на макаках. Профессор Мэри Герберт из британского Ньюкасла добилась разрешения провести первую такую процедуру в 2017 году. Но Джон Чжан, потерпев фиаско в Китае с починкой бесплодия, все-таки успел быстрее всех.
Профессор Мэри Герберт из британского Ньюкасла добилась разрешения провести первую такую процедуру в 2017 году. Но Джон Чжан, потерпев фиаско в Китае с починкой бесплодия, все-таки успел быстрее всех.
Джон Чжан с первым ребенком от трех родителей
New Hope Fertility Center
Первый «его» ребенок появился на свет в Мексике в 2016 году, где власти регулированием деторождения не столь озабочены. Родители мальчика были мусульманами, и классический метод переноса пронуклеусов для них был невозможен — для этого пришлось бы разрушить оплодотворенную яйцеклетку донора, то есть убить зародыш, что религиозные нормы родителей не позволяли. Поэтому Чжан использовал альтернативный метод — перенос веретена, то есть сначала пересадил генетический материал матери в донорскую яйцеклетку (без ядра), а затем устроил ей «свидание» со сперматозоидом отца. Но и такой трюк не пришелся мировой общественности по вкусу. Родившегося мальчика окрестили «ребенком от трех родителей», и начался новый скандал.
Двери закрываются
Одни ученые обвинили Чжана в экспериментах на живых людях, другие предложили проводить подобные испытания только на эмбрионах мужского пола, которые заведомо не передадут «результат» эксперимента потомству. Третьи задались вопросом: есть ли у Чжана доказательства того, что у ребенка не возникнет гетероплазмии или даже отката к изначальному состоянию? Доказательств у Чжана не было: родители забрали ребенка и отказались от долгосрочного наблюдения.
Итог скандала был предсказуем: FDA укрепило возведенную прежде «бумажную стену» и запретило любые манипуляции по замещению митохондрий. Великобритания осталась единственной страной, где они сейчас официально одобрены — в редких случаях и после долгих обсуждений наверху, в кабинетах Управления по оплодотворению человека и эмбриологии. Всем остальным желающим экспериментировать с яйцеклетками и их митохондриями приходится искать себе страну, где законодательство никак эту методику не регулирует, и не слишком сильно афишировать свои исследования.
Всем остальным желающим экспериментировать с яйцеклетками и их митохондриями приходится искать себе страну, где законодательство никак эту методику не регулирует, и не слишком сильно афишировать свои исследования.
Митохондриальные болезни могли бы стать первыми генетическими болезнями, которые люди научились лечить массово — но не стали. К методике митохондриального переноса прочно приклеилось название «ребенок от трех родителей», и несмотря на то, что сами исследователи считают его некорректным — донорских генов всего 37, а от отца и матери их по 20 тысяч — оно теперь устойчиво ассоциируется с нарушением этических норм. Поэтому, чтобы решить проблему бесплодия или избавить своего ребенка от риска стать обладателем целого букета неизлечимых болезней, родителям приходится отправляться в «эмбриологические турне», иногда на другой край света.
ЭМ-снимок митохондрии. Черные точки близко к поверхности мембраны — это мтДНК, помеченная частицами золота
Francisco J Iborra et al. / BMC Biology, 2004 / CC BY 2.0
А потом появился способ вылечить генетические болезни, скрытые уже не в органеллах клетки, а прямо в ее ядре. Несмотря на то, что люди, которые первыми придумали применять CRISPR/Cas9 к человеческим генам, заранее предупреждали, что система к этому еще не готова, история повторилась. Воспользовавшись тем, что китайское законодательство закрыло калитку для манипуляций митохондриями, но ничего не сказало о редактировании генов, очередной первопроходец Цзянькуй Хэ опробовал CRISPR на эмбрионах. Дальше случилось то же, что и всегда: скандал, запреты, попытки не допустить повторения ситуации с «детьми от трех родителей» (впрочем, ВОЗ вот уже год с небольшим работает над стандартами надзора за манипуляциями с человеческим геномом, и упорно избегает слова «мораторий»; тем временем во многих странах официального запрета на CRISPR-детей нет до сих пор).
Но поскольку лечить генетические болезни все-таки нужно, появился компромиссный вариант — CRISPR-терапия. Иными словами, пока мир разбирается с тем, имеем ли мы право редактировать эмбрионов, можно тренироваться на взрослых: вводить им в кровь систему редактирования и чинить поломки прямо в работающих тканях. Этот метод уже отработали на самых разных клетках, и недавно перешли к испытаниям in vivo.
По мере того, как CRISPR отвоевывал себе одну терапевтическую область за другой, стало понятно, что против митохондриальных мутаций он бессилен. Дело в том, что большинство систем генетического редактирования работают, как ножницы, разрезая ДНК в условленном месте. И если ядерную ДНК после такого клетка легко восстанавливает, соединяя концы разрыва, то митохондриальную разрушает — в норме она свернута в кольцо, так что двунитевой разрыв считается не рядовой поломкой, а признаком серьезной проблемы. Поэтому потери от такого редактирования могут превысить выигрыш.
Так митохондриальные болезни не только не стали первым достижением генетической терапии, но и вовсе остались последним не взятым бастионом.
Параллельные дороги
Справедливости ради стоит сказать, что модификация эмбрионов — не единственный способ справиться с митохондриальными дефектами. Например, митохондрии можно пересаживать не в яйцеклетку, а в уже родившийся организм (подобно тому как сейчас вводят CRISPR/Cas).
Сейчас клинические испытания проходят две терапии такого рода. В рамках первой — наращивания митохондрий (mitochondrial augmentation therapy) — ребенок получает донорские митохондрии от матери (в случае, если его митохондриальная болезнь возникла с нуля, а не досталась от матери). У ребенка забирают клетки — например, стволовые клетки крови — и культивируют их вместе с митохондриями, выделенными из клеток матери. Считается, что при этом клетки крови ребенка поглощают материнские органеллы, становятся более жизнеспособными и будут активно размножаться после возвращения в организм, таким образом поддерживая работу «сломанных».
Вторая терапия предполагает, что ребенок становится донором митохондрий сам для себя — например, в случае ишемии сердца при родах или в первые часы жизни. Тогда из какой-нибудь скелетной мышцы вырезают кусочек ткани, выделяют оттуда митохондрии и вводят их в сердечную мышцу. Этот метод недавно опробовали на пяти новорожденных: двоих из них спасти не удалось, а еще трое выздоровели, но неизвестно, какую роль в этом сыграла митохондриальная аутотрансплантация.
Можно представить себе, что комбинация этих двух методов могла бы породить полноценную терапию, в ходе которой донорские митохондрии вводили бы в кровь пациента, а они заселяли бы поврежденные митохондриальной болезнью ткани. Однако у научного сообщества остается немало вопросов к этим процедурам. Несмотря на то, что отдельные митохондрии действительно могут выжить в плазме крови, неизвестно, способны ли клетки тела их захватывать, а если да, то выживают ли они внутри. Защитники метода отмечают, что «иногда необходимо принять технологию, даже если мы не знаем, как она работает».
Есть и более радикальные решения митохондриальных проблем: так, еще несколько лет назад самый знаменитый борец со старением Обри ди Грей предложил перенести все гены из митохондрии в ядро. Два из них его коллегам удалось переместить и показать, что даже оттуда они успешно справлялись со своими обязанностями.
И хотя этот проект кажется еще менее реалистичным, чем все прочие, может оказаться, что некоторые митохондриальные гены можно пересаживать по отдельности — подобно тому как с мутациями в ядерной ДНК пытаются справиться с помощью генной терапии. Такие работы тоже есть, есть и первые клинические испытания — так пытаются лечить наследственную оптическую нейропатию. Хитрость здесь в том, что генная терапия доставляет митохондриальный ген не в митохондрию, а в ядро. Тем не менее, можно так сконструировать искусственный ген, чтобы получившийся продукт клетка транспортировала в митохондрию, и тогда неважно, где он производится.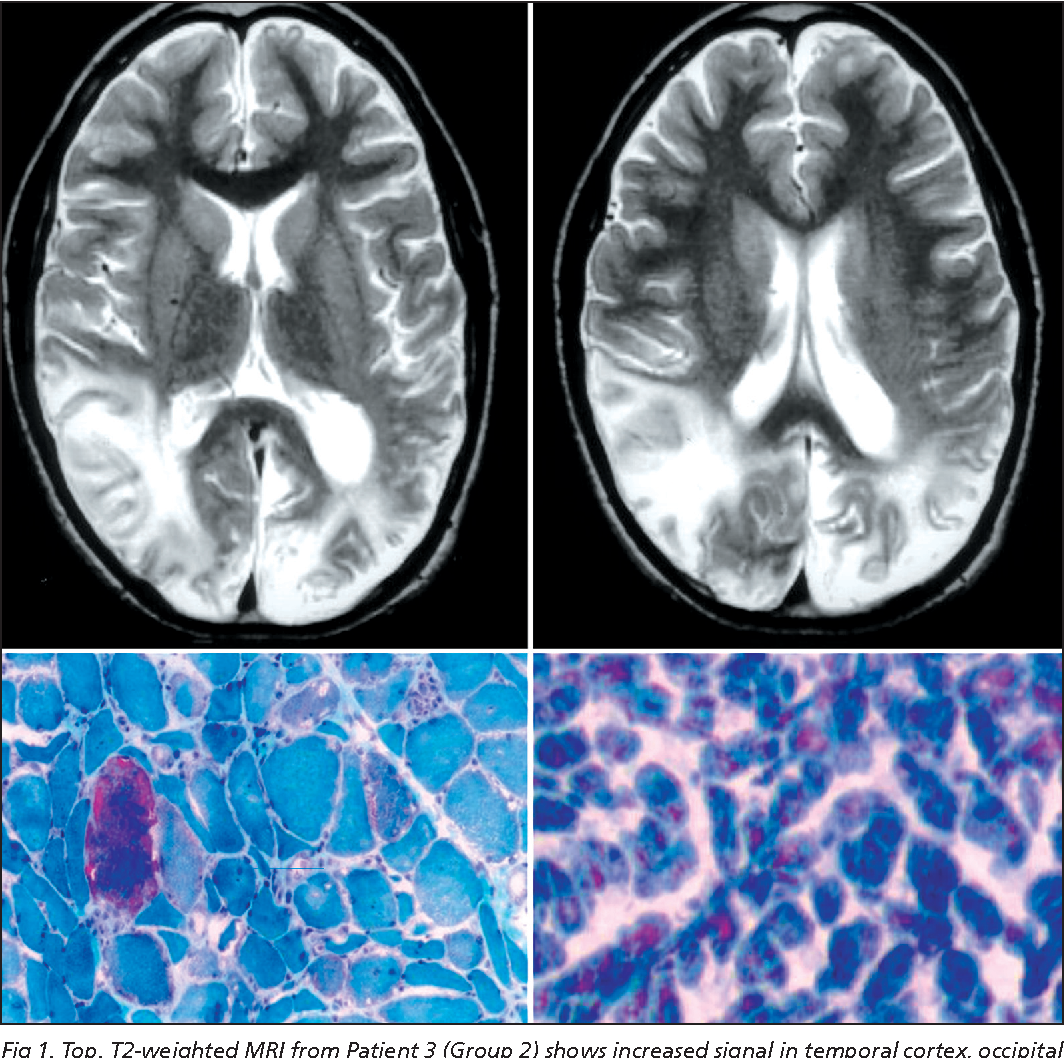
Новая тропа
И все же гораздо надежнее было бы переписать мутантный митохондриальный ген раз и навсегда. Этой задачей занялся Дэвид Лю, один из главных специалистов в мире по редактированию генома. Именно он в 2016 году придумал, как исправлять мутации, не разрезая ДНК, — и собрал редактор оснований (base editor). Это молекулярная система из двух ферментов: dCas9, который наводится на конкретное место в ДНК, и дезаминазы, что исправляет один нуклеотид на другой, буквально переписывая «генетический текст» наживую.
Для митохондрий и этот метод не годится. Редакторы оснований напрямую зависят от направляющей РНК, которая доставляет их к цели: потом Cas расплетает спираль ДНК на две отдельные нити, с одной связывается РНК, а другую атакует дезаминаза. Но направляющая РНК не может проникнуть внутрь митохондрии — не хватает транспортной системы, которая бы протаскивала ее сквозь две мембраны . Нужно было придумать какую-то систему, которая работает без РНК. Недавно команда Лю создала такую систему. И работает она еще и без Cas.
Система построена на основе антибиотика DddA, который выделяет бактерия Burkholderia cenocepacia. У него есть две важные особенности: во-первых, он действует точечно: исправляет в целевом гене все С (цитозиновые нуклеотиды) на А (адениновые) — точнее, сначала, переводит С в U (урацил), а клетка превращает их в А — то есть работает дезаминазой. Во-вторых, в отличие от всех других редакторов оснований, он связывается с двухцепочечной ДНК — а значит, нет необходимости ее разделять на две нити с помощью направляющей РНК, которая не пролезает в митохондрии.
Но просто так без направляющей РНК все равно не обойтись — необходим какой-то другой механизм, чтобы нацелить DddA на нужное место в митохондриальном геноме. И здесь команда Лю сделала шаг назад и воспользовалась технологией, которая, казалось бы, давно уступила место CRISPR — TALEN. Это бактериальные ферменты-конструкторы: они построены из доменов, каждый из которых распознает определенную последовательность ДНК. Подбирая нужный комплект доменов, можно добиться того, чтобы фермент садился на конкретное место в геноме. Эта технология, которая давно считается более сложной и дорогой, теперь может закрыть ту нишу, которая CRISPR оказалась не по зубам.
Подбирая нужный комплект доменов, можно добиться того, чтобы фермент садился на конкретное место в геноме. Эта технология, которая давно считается более сложной и дорогой, теперь может закрыть ту нишу, которая CRISPR оказалась не по зубам.
Соединив подобранный TALEN с нетоксичной частью DddA (той, что способна только дезаминировать ДНК, а не распознавать ее участки), команда Лю получила заветный инструмент. Правда, для клинического применения он еще сыроват: в разных экспериментах он смог переписать не больше половины своих мишеней в клетках. Тем не менее, он проникает в митохондрии и не разрушает их изнутри, и это гораздо важнее, чем эффективность, которую несложно нарастить.
И если это удастся сделать, то мы сможем считать, что в организме человека больше нет такого гена, который мы не в силах изменить. Не останется ни единого участка ДНК, который будет нам неподвластен.
Инструмент Лю не требует никакого «третьего родителя», а его работа даже отдаленно не напоминает клонирование. А значит, шансы на то, чтобы оказаться не задевающим ничьи чувства и не вызывающим оправданные опасения, у него выше. Но каков будет следующий поворот этого сюжета? Вариантов два: долгие и тщательные испытания и постепенное применение нового редактора на взрослых людях (как происходит, например, с генной терапией) или авантюра с участием эмбрионов и попытки очередного первопроходца опередить свое время (так было с митохондриальной трансплантацией, так было с CRISPR и как, возможно, будет еще не раз). До клиники митохондриальному редактору еще далеко. Но делать ставки на то, какая судьба его ждет через несколько лет, можно уже сейчас: будет ли это очередной скандал, запрет и поиск новой дороги — или же в конце этой истории все-таки можно будет поставить, наконец, точку вместо вопросительного знака?
Полина Лосева
Митохондриальная болезнь | Бостонская детская больница
Что такое митохондриальная болезнь?
Митохондриальная болезнь — это не отдельное заболевание, а обобщающий термин для десятков отдельных заболеваний, при которых клетки организма имеют проблемы с выработкой энергии. В совокупности эти нарушения затрагивают от 1 из 6000 до 1 из 8000 живорождений, что делает митохондриальные заболевания почти такими же распространенными, как рак у детей. Однако по отдельности эти состояния очень редки.Митохондриальные нарушения часто называют митохондриальными энцефаломиопатиями и включают в себя следующие состояния, а также многие другие:
В совокупности эти нарушения затрагивают от 1 из 6000 до 1 из 8000 живорождений, что делает митохондриальные заболевания почти такими же распространенными, как рак у детей. Однако по отдельности эти состояния очень редки.Митохондриальные нарушения часто называют митохондриальными энцефаломиопатиями и включают в себя следующие состояния, а также многие другие:
- Синдром Кирнса-Сайра
- Болезнь Ли
- MELAS (митохондриальная энцефаломиопатия, лактоацидоз и приступы, похожие на инсульт)
- MERRF (миоклоническая эпилепсия с рваными красными волокнами)
- MNGIE (митохондриальная нейрогастроинтестинальная энцефалопатия)
- Синдром Пирсона
- нарушения обмена пирувата, такие как недостаточность пируватдегидрогеназы
- нарушения цикла Кребса
Митохондриальные нарушения являются генетическими и иногда передаются в семье.Они могут вызывать широкий спектр симптомов, от задержки развития и потери слуха до судорог, инсультов, сердечной недостаточности и диабета в различных комбинациях. Для этих заболеваний типично поражение нескольких органов. Заболевание может варьироваться от тяжелого, начинающегося при рождении, до легкого, которое не проявляется до зрелого возраста. Иногда это может появиться внезапно, когда ребенка бросает вызов другой болезни.
В настоящее время нет лекарства от митохондриальной болезни. Лечение состоит в основном из поддерживающей терапии, а также некоторых витаминов и добавок.Лечение подбирается индивидуально в зависимости от митохондриального нарушения и симптомов у ребенка. Поскольку дети с митохондриальными заболеваниями очень чувствительны даже к незначительным заболеваниям и другим факторам стресса, важно внимательно следить за общим состоянием здоровья вашего ребенка и тесно сотрудничать со специалистами, чтобы обеспечить наилучший уход за «всем ребенком».
Чтобы узнать больше о том, как мы лечим митохондриальные заболевания в Boston Children’s, посетите страницу Митохондриальной программы.
Митохондриальная болезнь | Детская больница Филадельфии
Митохондриальное заболевание, или митохондриальное заболевание, относится к группе заболеваний, которые влияют на митохондрии, которые представляют собой крошечные компартменты, присутствующие почти в каждой клетке тела.Основная функция митохондрий — вырабатывать энергию. Для производства большего количества энергии необходимо больше митохондрий, особенно в органах с высоким потреблением энергии, таких как сердце, мышцы и мозг. Когда количество или функция митохондрий в клетке нарушается, вырабатывается меньше энергии и возникает дисфункция органов.
В зависимости от того, какие клетки в организме разрушили митохондрии, могут возникать разные симптомы. Митохондриальные заболевания могут вызывать широкий спектр проблем со здоровьем, включая усталость, слабость, метаболические инсульты, судороги, кардиомиопатию, аритмии, нарушения развития или когнитивные нарушения, сахарный диабет, нарушение слуха, зрения, роста, функции печени, желудочно-кишечного тракта или почек и более.Эти симптомы могут проявляться в любом возрасте от младенчества до позднего взросления.
Каждые 30 минут рождается ребенок, у которого к 10 годам разовьется митохондриальное заболевание. В целом, примерно у 1 из 4 300 человек в Соединенных Штатах есть митохондриальное заболевание. Учитывая различные потенциальные проявления, которые могут возникнуть, митохондриальное заболевание может быть трудным для диагностики и часто неправильно диагностируется.
Существуют различные методы определения наличия у человека митохондриальных заболеваний.К ним относятся генетические диагностические тесты, генетические или биохимические тесты в пораженных тканях, таких как мышцы или печень, и другие биохимические маркеры крови или мочи. Однако наши знания все еще растут, и мы еще не знаем все гены, которые потенциально могут вызывать митохондриальные заболевания.
Митохондрии уникальны тем, что имеют собственную ДНК, называемую митохондриальной ДНК или мтДНК. Мутации в этой мтДНК или мутации в ядерной ДНК (ДНК, обнаруженной в ядре клетки) могут вызывать митохондриальные нарушения.Токсины из окружающей среды также могут вызывать митохондриальные заболевания.
Симптомы митохондриального расстройства включают:
- Слабый рост
- Нарушение координации мышц, мышечная слабость
- Проблемы неврологического характера, включая судороги
- Расстройство аутистического спектра, представленное различными характеристиками РАС
- Проблемы со зрением и / или слухом
- Задержки в развитии, нарушения обучаемости
- Болезни сердца, печени или почек
- Желудочно-кишечные расстройства, такие как тяжелые запоры
- Диабет
- Нарушения органов дыхания
- Повышенный риск заражения
- Дисфункция щитовидной железы и / или надпочечников
- Дисфункция вегетативной нервной системы
- Нейропсихологические изменения или деменция, характеризующаяся спутанностью сознания, дезориентацией и потерей памяти
В настоящее время не существует высокоэффективного лечения митохондриального заболевания.Лечение митохондриальных заболеваний — это поддерживающая терапия, которая может включать в себя режим питания, упражнения и / или витаминные или аминокислотные добавки.
Знание первопричины вашего состояния или состояния вашего ребенка поможет вашей медицинской бригаде выбрать лучший курс лечения. Митохондриальная медицина в Детской больнице Филадельфии (CHOP) тесно сотрудничает с вашим лечащим врачом, неврологом и другими специалистами, чтобы удовлетворить ваши повседневные медицинские потребности.
Наша команда предоставит соответствующие консультации по митохондриальным заболеваниям на основе диагноза вашего ребенка, включая обзор особенностей митохондриальных заболеваний и генетики.
Невролог или терапевт вашего ребенка решит повседневные медицинские проблемы, связанные с митохондриальными заболеваниями.
В долгосрочном уходе могут участвовать различные специалисты, включая неврологов, эндокринологов, офтальмологов, аудиологов, кардиологов и нефрологов.
Пациентам с митохондриальными заболеваниями без известной генетической причины обычно рекомендуются ежегодные контрольные визиты, поскольку мутации в новых генах, вызывающих митохондриальные заболевания, обнаруживаются постоянно.
Митохондриальных заболеваний | Детская больница CS Mott
Обзор темы
Что такое митохондриальные заболевания?
Митохондрии (скажем «my-tuh-KAWN-dree-uh») — это крошечные части клеток вашего тела. Митохондрии часто называют электростанцией клетки, потому что одна из их задач — производить энергию.
Митохондриальные болезни — это группа редких заболеваний, которые могут передаваться от матери к детям. Заболевания возникают, когда митохондрии не работают должным образом.
Заболевания могут появиться в любом возрасте. И они могут поражать множество различных участков тела.
Примеры этих заболеваний включают:
- Синдром Кирнеса-Сейра. Симптомы обычно появляются в возрасте до 20 лет и поражают глаза и уши.
- Синдром Ли. Поражает нервную систему малыша. Обычно это начинается в первый год жизни.
- Синдром MERRF. MERRF влияет на нервную систему, мышцы и другие части тела.
- MELAS-синдром. MELAS влияет на многие участки тела, включая мозг, нервную систему и мышцы.
Каковы симптомы?
Симптомы зависят от типа клеток, повредивших митохондрии. Симптомы могут варьироваться от человека к человеку.
Митохондриальная болезнь клеток головного мозга может вызывать такие симптомы, как:
- Снижение умственных способностей.
- Изъятия.
- штрихов.
Когда болезнь поражает мышечных клеток , симптомы могут включать:
- Слабость.
- Судороги.

- Проблемы с движением.
В клетках ушей или глаз митохондриальные заболевания могут вызывать:
- Глухоту.
- Слепота.
- Веки отвисшие.
- Проблемы с движением глаз.
Другие части тела, которые могут быть поражены, включают почки, нервы или печень.
Как диагностируются митохондриальные заболевания?
Если вы или ваш врач считаете, что у вас митохондриальное заболевание, ваш врач проведет медицинский осмотр и медицинские тесты. Ваш врач задаст вопросы о вас и вашем здоровье. Он или она также спросит о здоровье вашей семьи, чтобы узнать, есть ли у кого-нибудь в вашей семье подобные симптомы.
Тип медицинского обследования, который вы пройдете, будет зависеть от ваших симптомов и от того, какие части вашего тела затронуты. Тесты, которые могут быть выполнены, включают биопсию мышц и визуализационные тесты, такие как МРТ.Ваш врач может также попросить вас пройти генетический тест.
Ваш врач может посоветовать вам обратиться к генетическому консультанту. Эти консультанты обучены объяснять генетический тест и его результаты, но решение о том, проходить ли этот тест, принимаете вы. Они также предоставляют образование и поддержку семьям, члены которых имеют врожденные дефекты или генетические заболевания, такие как митохондриальные заболевания. Генетическое консультирование может помочь вам понять ваш риск рождения ребенка с митохондриальным заболеванием.
Как к ним относятся?
Лечение обычно направлено на облегчение симптомов и улучшение самочувствия.Вы и ваш врач будете работать вместе, чтобы найти наиболее подходящее для вас лечение. Возможно, вам понадобится обратиться к специалисту или группе специалистов.
Лечение может включать:
- Лечебная физкультура.
- Употребление здоровой пищи.
- Достаточно отдыхать.
- Избегать больных.
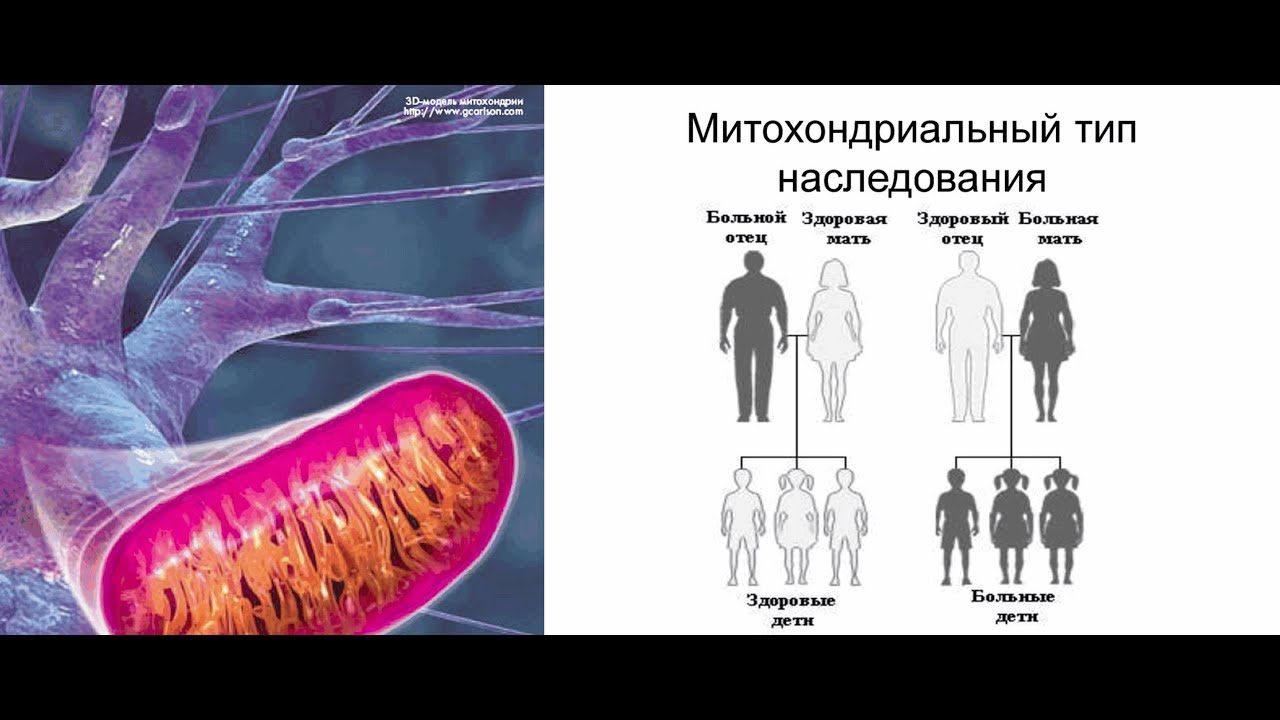
- Поддержание комфортной температуры.
Ваш врач также может порекомендовать принимать определенные витамины и добавки.
Хирургия или другие процедуры могут быть рекомендованы при таких физических симптомах, как опущение век, катаракта или потеря слуха.
Часто задаваемые вопросы о митохондриальных заболеваниях
Q: Что такое митохондриальные заболевания или нарушения?
A: Митохондрии — это крошечные части почти каждой клетки вашего тела. Митохондрии подобны электростанции клеток. Они превращают сахар и кислород в энергию, необходимую клеткам для работы.
При митохондриальных заболеваниях митохондрии не могут эффективно превращать сахар и кислород в энергию, поэтому клетки не работают правильно.
Существует много типов митохондриальных заболеваний, и они могут поражать разные части тела: мозг, почки, мышцы, сердце, глаза, уши и другие. Митохондриальные заболевания могут поражать одну часть тела или многие части. Они могут повлиять на эти части в легкой или очень серьезной степени.
Не у всех митохондриальных заболеваний проявляются симптомы. Однако при обсуждении группы митохондриальных заболеваний, которые имеют тенденцию поражать детей, симптомы обычно проявляются в раннем и дошкольном возрасте.
Митохондриальные заболевания и нарушения — это одно и то же.
В: Есть ли связь между митохондриальными заболеваниями и аутизмом?
A: Ребенок с митохондриальной болезнью:
- также может иметь расстройство аутистического спектра,
- может иметь некоторые из симптомов / признаков аутизма, или
- может не иметь никаких признаков или симптомов, связанных с аутизмом.
Ребенок с аутизмом может иметь или не иметь митохондриальное заболевание.Когда у ребенка аутизм и митохондриальное заболевание, у него иногда возникают и другие проблемы, включая эпилепсию, проблемы с мышечным тонусом и / или двигательные нарушения.
Необходимы дополнительные исследования, чтобы выяснить, насколько часто люди страдают аутизмом и митохондриальными нарушениями. Сейчас это кажется редкостью. В целом, необходимы дополнительные исследования митохондриальных заболеваний и аутизма.
В: Что такое регрессивная энцефалопатия?
A: Энцефалопатия — это медицинский термин, обозначающий заболевание или расстройство головного мозга.Обычно это означает замедление работы мозга.
Регресс происходит, когда человек теряет навыки, которые у него были раньше, такие как ходьба, разговор или даже общение.
Регрессивная энцефалопатия означает заболевание или расстройство мозга, из-за которого человек теряет навыки, которыми он когда-то обладал.
Мы знаем, что иногда кажется, что дети с митохондриальными заболеваниями развиваются должным образом, но в раннем или дошкольном возрасте они регрессируют. Болезнь была все время, но случается что-то, что ее «запускает».Это может быть что-то вроде недоедания, такого заболевания, как грипп, высокая температура, обезвоживание, или что-то еще.
В: Есть ли связь между аутизмом и энцефалопатией?
A: Большинство детей с расстройством аутистического спектра не страдают и не болели энцефалопатией. У некоторых детей с расстройством аутистического спектра наблюдался регресс, а у некоторых — регрессивная энцефалопатия.
Вопрос: Что мы знаем о связи между митохондриальными заболеваниями и другими нарушениями, связанными с мозгом?
A: Различные части мозга выполняют разные функции.Область мозга, поврежденная митохондриальной болезнью, определяет, как это повлияет на человека. Это означает, что у человека могут быть судороги; проблемы с разговором или взаимодействием с людьми; трудности с приемом пищи; мышечная слабость или другие проблемы. У них может быть одна проблема или несколько.
В: Вызывают ли вакцины митохондриальные заболевания или усугубляют их?
A: На данный момент нет научных исследований, которые утверждали бы, что вакцины вызывают или усугубляют митохондриальные заболевания. Мы знаем, что некоторые заболевания, которые можно предотвратить с помощью вакцин, например грипп, могут вызвать регресс, связанный с митохондриальным заболеванием. Необходимы дополнительные исследования, чтобы определить, есть ли редкие случаи, когда основные митохондриальные нарушения вызываются чем-либо, связанным с вакцинами. Однако мы знаем, что для большинства детей вакцины — это безопасный и важный способ предотвратить заражение опасными для жизни заболеваниями.
Мы знаем, что некоторые заболевания, которые можно предотвратить с помощью вакцин, например грипп, могут вызвать регресс, связанный с митохондриальным заболеванием. Необходимы дополнительные исследования, чтобы определить, есть ли редкие случаи, когда основные митохондриальные нарушения вызываются чем-либо, связанным с вакцинами. Однако мы знаем, что для большинства детей вакцины — это безопасный и важный способ предотвратить заражение опасными для жизни заболеваниями.
В: Все ли дети проходят стандартное тестирование на митохондриальные заболевания? А как насчет детей с аутизмом?
A: Обычно детей не тестируют на митохондриальные заболевания.Сюда входят дети с аутизмом и другими задержками в развитии.
Тестирование — непростая задача, требующая взятия нескольких образцов крови, а часто и образцов мышц. Врачи решают, следует ли проводить тестирование на митохондриальные заболевания, основываясь на признаках и симптомах ребенка.
В: Следует ли мне сдать анализ на митохондриальные заболевания у моего ребенка?
A: Если вы беспокоитесь, что у вашего ребенка может быть митохондриальное заболевание, поговорите с врачом вашего ребенка.
вопросов и ответов — Фонд Лили для исследований митохондриальных заболеваний и других метаболических нарушений
Донорство яйцеклеток
Это включает использование яйцеклетки женщины-донора в сочетании со спермой отца.
Это лечение эффективно как при дефектах митохондриальной ДНК, так и при дефектах ядерной ДНК, и гарантирует, что в результате этого ребенок будет свободен от митохондриальной болезни.
Ребенок будет генетически связан с отцом, но не с матерью.
Донорство спермы
Это включает использование спермы мужчины-донора в сочетании с яйцеклеткой матери.
Это лечение эффективно только при дефектах ядерной ДНК, но гарантирует, что у полученного ребенка не будет митохондриальной болезни.
Ребенок будет генетически связан с матерью, но не с отцом.
ЭКО с PGD
Этот метод включает лечение ЭКО с использованием материнской яйцеклетки и отцовской спермы, поэтому полученный ребенок будет генетически связан с обоими родителями.Процесс включает создание ряда эмбрионов с помощью обычного процесса ЭКО, а затем их скрининг с использованием процесса, называемого преимплантационной генетической диагностикой (ПГД), для выявления эмбрионов, которые выглядят здоровыми и либо свободны от болезней, либо имеют низкий уровень мутации.
Процесс скрининга очень эффективен в отношении мутаций ядерной ДНК, поскольку может дать однозначный ответ «да» или «нет».
Однако результаты мутаций митохондриальной ДНК могут быть менее ясными. Клиницисты обычно могут посоветовать семьям процент дефектной митохондриальной ДНК, который затем сравнивается с теоретическими пороговыми значениями заболевания.Однако пограничные результаты означают, что семьи могут не получить необходимых гарантий, чтобы забеременеть.
Для некоторых женщин, у которых есть «гомоплазматические мутации», этот метод не будет успешным, и для этих семей, а также для семей, где ПГД не увенчалась успехом, недавно произошел прорыв в медицине под названием «Донорство митохондрий».
Донорство митохондрий
Этот метод включает использование материнской яйцеклетки, отцовской спермы и здоровых митохондрий от женщины-донора.В результате ребенок будет генетически связан с обоими родителями и будет иметь крошечные 0,02% митохондриальной ДНК от женщины-донора.
Это лечение эффективно только для матерей с дефектами митохондриальной ДНК, но оно может помочь некоторым семьям, которые ЭКО с ПГД не могут.
В феврале 2015 г. было принято законодательство Великобритании
, позволяющее предлагать эту технику на индивидуальной основе подходящим пострадавшим семьям.
Я хочу узнать больше о донорстве митохондрий
Вернуться наверх
Митохондриальные миопатии — Фонд детской неврологии
Описание
Митохондриальные миопатии — это группа нервно-мышечных заболеваний, вызываемых повреждением митохондрий — небольших, производящих энергию структур, которые служат «электростанциями» клеток. «Нервным клеткам в головном мозге и мышцах требуется много энергии, и поэтому они, по всей видимости, особенно повреждаются при митохондриальной дисфункции. Некоторые из наиболее распространенных митохондриальных миопатий включают синдром Кернса-Сейра, миоклоническую эпилепсию с рваными красными волокнами и митохондриальные миопатии. энцефаломиопатия с лактоацидозом и эпизодами, напоминающими инсульт. Симптомы митохондриальной миопатии включают мышечную слабость или непереносимость физических упражнений, сердечную недостаточность или нарушения ритма, деменцию, двигательные расстройства, эпизоды, похожие на инсульт, глухоту, слепоту, опущенные веки, ограниченную подвижность глаз. , рвота и судороги.Прогноз для этих расстройств варьируется от прогрессирующей слабости до смерти. Большинство митохондриальных миопатий возникают в возрасте до 20 лет и часто начинаются с непереносимости физических упражнений или мышечной слабости. Во время физической активности мышцы могут быстро утомляться или слабеть. Мышечные спазмы возникают редко, но могут возникать. С этими расстройствами также связаны тошнота, головная боль и одышка.
«Нервным клеткам в головном мозге и мышцах требуется много энергии, и поэтому они, по всей видимости, особенно повреждаются при митохондриальной дисфункции. Некоторые из наиболее распространенных митохондриальных миопатий включают синдром Кернса-Сейра, миоклоническую эпилепсию с рваными красными волокнами и митохондриальные миопатии. энцефаломиопатия с лактоацидозом и эпизодами, напоминающими инсульт. Симптомы митохондриальной миопатии включают мышечную слабость или непереносимость физических упражнений, сердечную недостаточность или нарушения ритма, деменцию, двигательные расстройства, эпизоды, похожие на инсульт, глухоту, слепоту, опущенные веки, ограниченную подвижность глаз. , рвота и судороги.Прогноз для этих расстройств варьируется от прогрессирующей слабости до смерти. Большинство митохондриальных миопатий возникают в возрасте до 20 лет и часто начинаются с непереносимости физических упражнений или мышечной слабости. Во время физической активности мышцы могут быстро утомляться или слабеть. Мышечные спазмы возникают редко, но могут возникать. С этими расстройствами также связаны тошнота, головная боль и одышка.
Лечение
Хотя не существует специального лечения митохондриальной миопатии, физиотерапия может расширить диапазон движений мышц и улучшить их ловкость.Витаминная терапия, такая как рибофлавин, коэнзим Q и карнитин (специализированная аминокислота), может обеспечить субъективное улучшение утомляемости и уровня энергии у некоторых пациентов.
Прогноз
Прогноз для пациентов с митохондриальной миопатией сильно различается в зависимости от типа заболевания и степени поражения различных органов. Эти расстройства вызывают прогрессирующую слабость и могут привести к смерти.
Исследования
NINDS проводит и поддерживает исследования митохондриальных миопатий. Цели этого исследования — расширить научное понимание этих расстройств и найти способы их эффективного лечения, предотвращения или потенциального лечения.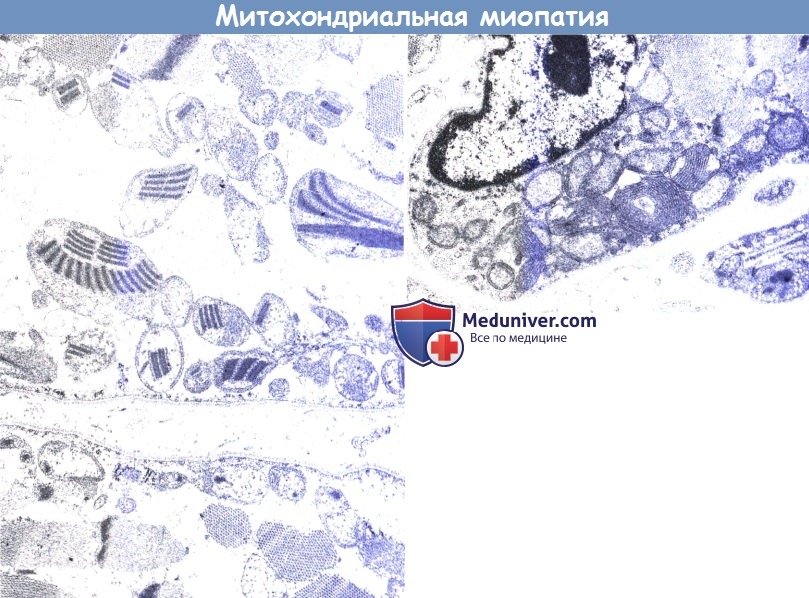
Информация из Национальной медицинской библиотеки MedlinePlusMetabolic Disorders
Информация получена в рамках партнерства CNF с Национальным институтом неврологических расстройств и инсульта (NINDS), Национальным институтом здравоохранения США .
Выявлена наиболее частая причина смерти от митохондриальной болезни у детей
Исследователи изучили факторы риска смертности у детей с митохондриальными заболеваниями и определили, что основными причинами смерти являются сепсис, инфекция крови и пневмония. В исследовании подчеркивается важность ранней диагностики и тщательного наблюдения за этими маленькими пациентами.
Статья «Причина смерти детей с митохондриальными заболеваниями» была опубликована в журнале Детская неврология.
Митохондриальные заболевания могут представлять собой множество различных состояний с множеством различных симптомов. Они вызваны дефектом клеточной органеллы, называемой митохондриями, которая производит энергию, которая поддерживает функционирование тела и его органов.
Эти дефекты митохондриальной функции могут возникать в любом органе, но, как правило, возникают в органах, которые используют много энергии, таких как мозг, сердце, печень и скелетные мышцы — мышцы, прикрепленные к костям.
Исследователи изучили истории болезни 221 педиатрического пациента с диагнозом митохондриальные заболевания и наблюдались в больнице Каннам Северанс Медицинского колледжа Университета Йонсей в Сеуле, Южная Корея, с января 2006 года по январь 2015 года.
Среди прочего были исследованы причины смерти, количество смертей, пол и время выживания.
Из 221 ребенка, включенного в исследование, 31 из них, или 14%, умер. Большинство умерших пациентов (23 или 75%) жили от трех до девяти лет; пять пациентов (16%) прожили менее трех лет; и трое пациентов (10%) жили более девяти лет.
Причинами смерти детей в возрасте до 15 лет с митохондриальными заболеваниями в основном были сепсис (17 пациентов, или 55%) и пневмония (13 пациентов, или 42%).