
Митохондриальная энцефалопатия что это такое: MELAS-синдром. Что такое MELAS-синдром?

MELAS-синдром. Что такое MELAS-синдром?
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
MELAS-синдром – это генетическое заболевание с поражением центральной нервной системы, мышечной ткани и различных органов. В основе патологии лежит нарушение тканевого дыхания и дефект энергетического метаболизма. Клиническая картина отличается гетерогенностью, включает острые эпизоды, напоминающие инсульт, эпилептические приступы, непереносимость физических нагрузок вследствие мышечной слабости. Для диагностики применяются МРТ, ЭЭГ, ЭНМГ и другие исследования. Окончательный диагноз устанавливается при обнаружении точечных мутаций в митохондриальной ДНК. Лечение заключается в назначении метаболических препаратов и симптоматической терапии.
МКБ-10
E88.8 G71.3
- Причины
- Патогенез
- Классификация
- Симптомы
- Осложнения
- Диагностика
- Лечение MELAS-синдрома
- Прогноз и профилактика
- Цены на лечение
Общие сведения
MELAS-синдром (митохондриальная энцефалопатия с лактат-ацидозом и инсультоподобными эпизодами) относится к болезням, обусловленным генетическим дефектом митохондриальной ДНК. При этом синдроме нарушается энергопродукция в митохондриальной дыхательной цепи. Заболевание впервые было описано в 1984 году. По разным данным, патология встречается с частотой от 1:15 000 до 1:20 000 человек. Гендерные различия отсутствуют. Средний возраст дебюта – 6-10 лет.
MELAS-синдром
Причины
Причиной возникновения патологии являются точечные мутации митохондриальной ДНК. На сегодняшний день известно около 10 генов, при дефекте которых наблюдается манифестация MELAS-синдрома. Наиболее часто выявляются мутации в генах, кодирующих транспортную РНК. Чаще всего (80-90% случаев) обнаруживается мутация A3243G в гене MTTL1 транспортной РНК аминокислоты лейцина.
Наиболее часто выявляются мутации в генах, кодирующих транспортную РНК. Чаще всего (80-90% случаев) обнаруживается мутация A3243G в гене MTTL1 транспортной РНК аминокислоты лейцина.
Из-за дефекта нарушается синтез митохондриальных белков. Наличие мутации необязательно гарантирует фенотипическое проявление заболевания, что связано с феноменом гетероплазмии, то есть, одновременного наличия в клетке, органе или организме нормальной и мутантной митохондриальных ДНК. Большое количество дефектных ДНК повышает вероятность клинической манифестации синдрома.
Патогенез
Генетически детерминированный дефект синтеза митохондриальных белков (ферментов тканевого дыхания) приводит к нарушению процессов окислительного фосфорилирования – последней стадии расщепления биологических субстратов: жирных кислот, углеводов. Окислительное фосфорилирование считается главным звеном энергетического метаболизма, обеспечивает образование подавляющего количества энергоносителей – молекул АТФ.
Сбой этих процессов происходит в каждой митохондрии с дефектной ДНК. Поскольку митохондрии представлены практически во всех клетках организма, поражение носит мультисистемный характер. Сильнее всего страдают органы и ткани с высокой энергетической потребностью: центральная и периферическая нервная система, миокард, скелетные мышцы.
В мышцах энергетический дефицит (тканевая гипоксия) приводит к избыточному образованию молочной кислоты. В генезе острых церебральных нарушений лежит дефицит оксида азота вследствие накопления в гладких мышцах сосудов фермента цитохромоксидазы. При недостаточности оксида азота возникает вазоконстрикция, агрегация тромбоцитов.
Классификация
По выраженности клинических проявлений выделяют 3 формы MELAS-синдрома:
- Бессимптомное носительство – наличие генетической мутации и изменений при биопсии мышц на фоне полного отсутствия клинических признаков заболевания.
- Олигосимптоматическая – обнаруживаются отдельные компоненты синдрома: мышечная слабость, головные боли и пр.

- Манифестная – яркая клиническая картина с острыми эпизодами.
Симптомы
Обычно вначале манифестируют неврологические симптомы, которые возникают уже в 6-15 лет. Наиболее типичными считаются инсультоподобные эпизоды (метаболический инсульт). Частые проявления – гемианопсия (выпадение половины поля зрения), нарушение равновесия, восприятия или воспроизведения речи, изменения сознания.
У половины пациентов отмечаются фокальные или генерализованные тонико-клонические эпилептические припадки, головные боли, напоминающие мигрень (односторонние, пульсирующие). Характерные черты таких приступов – возраст больных менее 40 лет, провоцирование инфекционным заболеванием, быстрый регресс симптоматики и частое рецидивирование.
Реже наблюдаются острые психозы и гемипарезы. Из других неврологических проявлений можно выделить задержку нервно-психического развития, заторможенность, когнитивные нарушения. Вследствие атрофии зрительных нервов постепенно ухудшается зрение. Еще один специфичный признак MELAS-синдрома – выраженная мышечная слабость и плохая переносимость физической нагрузки.
Вследствие атрофии зрительных нервов постепенно ухудшается зрение. Еще один специфичный признак MELAS-синдрома – выраженная мышечная слабость и плохая переносимость физической нагрузки.
Часто встречаются боли, судороги в мышцах. Мышечные спазмы также могут вызываться снижением концентрации кальция в крови вследствие гипопаратиреоза. У многих пациентов наблюдается постепенное ухудшение слуха (нейросенсорная тугоухость), возникает сахарный диабет. Кардиологические признаки заболевания включают кардиомиопатии, аритмии, хроническую сердечную недостаточность.
Осложнения
MELAS-синдром является тяжелым заболеванием, характеризующимся широким спектром неблагоприятных последствий. Наиболее частые осложнения, приводящие к летальному исходу, связаны с митохондриальной энцефалопатией. К ним относятся повторные «метаболические инсульты» и эпилептические статусы. Небольшая часть пациентов впадает в кому.
У больных повышен риск развития жизнеугрожающих нарушений ритма (желудочковая тахикардия, фибрилляция), остановки сердца. Нейросенсорная тугоухость и атрофия зрительного нерва могут привести к полной потере слуха и зрения. В единичных случаях возникает кишечная непроходимость, хроническая почечная и печеночная недостаточность.
Нейросенсорная тугоухость и атрофия зрительного нерва могут привести к полной потере слуха и зрения. В единичных случаях возникает кишечная непроходимость, хроническая почечная и печеночная недостаточность.
Диагностика
Больных с MELAS-синдромом курируют врачи-неврологи, педиатры. При общем осмотре обращает на себя внимание низкий рост, выраженная гипотония, гипотрофия мышц. Заподозрить заболевание позволяет сочетание миопатии с острыми неврологическими расстройствами у пациента молодого возраста. Дополнительное обследование, направленное на уточнение диагноза, включает:
- Лабораторные исследования. В биохимическом анализе крови определяется высокий уровень молочной кислоты, глюкозы и гликированного гемоглобина, уменьшение концентрации кальция. При анализе крови на гормоны обнаруживают снижение содержания соматотропного и паратиреоидного гормонов. В общем анализе мочи часто выявляется протеинурия.
- МРТ головного мозга. Повреждения мозга визуализируются как очаги неправильной формы с четкими контурами и слабо повышенной интенсивностью МР-сигнала.
 Отмечаются признаки объемного воздействия на субарахноидальное пространство. Нередко выявляются кальцинаты в базальных ганглиях с преимущественной локализацией в затылочной и теменной областях. При повторном проведении МРТ обнаруживается флюктуация или исчезновение очагов.
Отмечаются признаки объемного воздействия на субарахноидальное пространство. Нередко выявляются кальцинаты в базальных ганглиях с преимущественной локализацией в затылочной и теменной областях. При повторном проведении МРТ обнаруживается флюктуация или исчезновение очагов. - МР-спектроскопия. Магнитно-резонансная спектроскопия головного мозга показывает значительное снижение высокоэнергетических фосфорных соединений и увеличение концентрации лактата.
- ЭЭГ. На электроэнцефалограмме просматривается замедление основной активности, диффузные нарушения биоэлектрических процессов головного мозга, признаки островолновой активности, усиливающейся на фоне гипервентиляции.
- ЭНМГ. Электронейромиография подтверждает наличие неспецифических признаков – сокращение длительности потенциала и амплитуды двигательных единиц, блок и замедление проведения нервного импульса.
- Биопсия мышц. Наиболее типичные морфологические изменения – феномен «рваных красных волокон» (миофибриллы с большим числом пролиферирующих измененных митохондрий), склероз перимизия, регионарные некрозы.
 Отмечается положительная гистохимическая окраска на цитохромоксидазу и сукцинатдегидрогеназу.
Отмечается положительная гистохимическая окраска на цитохромоксидазу и сукцинатдегидрогеназу. - Исследование ДНК. Решающий диагностический тест для верификации синдрома MELAS. В лимфоцитах периферической крови чаще других выявляется мутация A3243G гена MTTL1.
Дифференциальный диагноз острых эпизодов проводят с ишемическими инсультами, субарахноидальным кровоизлиянием, острыми инфекционными менингоэнцефалитами. Миопатический синдром следует отличать от наследственных мышечных дистрофий, полимиозита. Патологию также нужно дифференцировать с другими митохондриальными заболеваниями.
Лечение MELAS-синдрома
Радикальное лечение не разработано. Все методы терапии направлены на улучшение состояния пациента и носят лишь паллиативный характер. Необходима диета с ограничением содержания углеводов. Диетическое питание требуется для снижения уровня глюкозы, негативно влияющей на параметры энергетического обмена, и для коррекции сахарного диабета. Применяются следующие медикаменты:
Применяются следующие медикаменты:
- Метаболические препараты. Для улучшения окислительного фосфорилирования в митохондриях назначается комплексное энерготропное лечение, включающее лекарства, способствующие переносу электронов в дыхательной цепи (коэнзим Q10, янтарная кислота), кофакторы ферментов энергетического обмена (рибофлавин, никотинамид), антиоксиданты (аскорбиновая кислота, токоферол).
- Предшественники и донаторы оксида азота. Во время инсультоподобного эпизода эффективными оказались медикаменты, повышающие уровень оксида азота в крови (L-аргинин, цитруллин), способствующие вазодилатации и улучшению микроциркуляции.
- Препараты для коррекции КЩР. Средства, снижающие уровень лактата в крови (корректоры лактат-ацидоза) используются только в остром периоде у пациентов с очень высоким содержанием в крови молочной кислоты. Их назначение требует большой осторожности из-за способности оказывать токсическое действие на нервную ткань. Возможно внутривенное введение гидрокарбоната натрия.

- Глюкокортикостероиды. Применение гормонов коры надпочечников (преднизолон) приводит к значительному регрессу неврологической симптоматики.
- Средства для коррекции гормональных нарушений. При развитии у сахарного диабета рекомендованы сахароснижающие препараты (метформин, глибенкламид), а при их неэффективности – инсулинотерапия. При возникновении гипопаратиреоза в схему лечения добавляют кальций и витамин Д.
- Противоэпилептические препараты. Широко используемые в эпилептологии производные вальпроевой кислоты строго противопоказаны, поскольку ингибируют энергетический метаболизм. Предпочтение отдается клоназепаму, ламотриджину, топирамату.
В зависимости от клинической картины назначают кардиотропные средства (бета-блокаторы, антиаритмики, ингибиторы АПФ), корректоры печеночной недостаточности (альбумин и медикаменты, подавляющие образование аммиака), сеансы гемодиализа. Следует максимально избегать применения препаратов, угнетающих митохондриальную функцию (барбитураты, хлорамфеникол, статины), поскольку это приводит к ухудшению состояния больного.
Прогноз и профилактика
Синдром MELAS – тяжелое прогрессирующее заболевание с высокой частотой летальных исходов. Продолжительность жизни у людей с бессимптомным носительством и олигосимптоматической формой не отличается от таковой в общей популяции. При манифестной форме, по различным данным, срок жизни составляет от 7 до 40 лет с момента дебюта болезни.
Методы первичной профилактики не разработаны. Поскольку неврологические приступы достаточно часто провоцируются острой инфекцией, для их предотвращения рекомендуется постановка прививок от гриппа и меры общей профилактики, направленные на повышение сопротивляемости организма (регулярные физические нагрузки, закаливание).
Вы можете поделиться своей историей болезни, что Вам помогло при лечении болезни «MELAS-синдром».
Источники
- Митохондриальная энцефалопатия с инсультоподобными эпизодами и лактат-ацидозом (синдром MELAS): критерии диагностики, особенности эпилептических приступов и подходы к лечению на примере клинического случая/ Ямин М.
 А., Черникова И.В., Арасланова Л.В., Шевкун П.А.// Неврология, нейропсихиатрия, психосоматика – 2017 – №9.
А., Черникова И.В., Арасланова Л.В., Шевкун П.А.// Неврология, нейропсихиатрия, психосоматика – 2017 – №9. - Неврологические нарушения при митохондриальной энцефаломиопатии — лактат-ацидозе с инсультоподобными эпизодами (синдроме MELAS)/ Харламов Д.А., Крапивкин А.И., Сухоруков В.С., Куфтина Л.А., Грознова О.С.// Российский вестник перинатологии и педиатрии – 2012 — №4.
- Инсультоподобное течение митохондриальной энцефаломиопатии (синдром MELAS)/ Смирнова И.Н., Кистенёв Б.А., Кротенкова М.В., Суслина З.А.// Нервные болезни – 2006 — №1.
- Инсульты при митохондриальных заболеваниях/ Пизова Н.В.// Неврология, нейропсихиатрия, психосоматика – 2017 — №9.
- Настоящая статья подготовлена по материалам сайта: https://www.krasotaimedicina.ru/
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Митохондриальная энцефалопатия
См. фотогалерею статьи
Пациентка Г., 34 лет, впервые госпитализирована в 1невр. отд. в сентябре 2010. Жалобы при поступлении на судорожные генерализованные приступы в вечернее и ночное время, эпизоды потери сознания днём, на мышечные подёргивания лица, судороги в ногах, снижение толерантности к физическим нагрузкам, снижение памяти, нарушение навыков чтения и письма, нарушение речи. Анамнез заболевания: С 13 лет у пациентки появились приступы с потерей сознания и клонико-тоническими судорогами, через несколько месяцев присоединились миоклонические судороги с вовлечением мышц рук, ног, лица, которые были расценены как эпилепсия. На фоне приёма противосудорожных препаратов генерализованные приступы стали редкими, а миоклонии более частыми и генерализованными. В возрасте 16 и 17 лет развиваются приступы с нарушением глотания, обильным слюнотечением, нарушением речи и выраженной мышечной слабостью. Со слов матери при МРТ головного мозга выявлены очаги демиелинизации, был поставлен диагноз — рассеянный энцефаломиелит, проводились курсы плазмофереза и гормонотерапии, без эффекта. Пациентка лечилась в США, где клинико-морфологическая картина также была расценена как демиелинизирующий процесс. В 20 лет развился пароксизм с выраженными длительными и генерализующимися миоклониями. Проведено генетическое обследование, биопсия бедренной мышцы, при которой выявлены: Атрофия отдельных мышечных волокон, преимущественно 11 типа, изменение нервно-мышечных пластинок с исчезновением аксонального их компонента, при реакции на SDH только в единичных мышечных волокнах выявляется периферическая зона усиления реакции, свидетельствующая о скоплении митохондрий по периферии мышечного волокна, выявлены признаки миопатических изменений в виде нарушения архитектуры и очагового лизиса миофибриллярного аппарата, в части волокон — признаки метаболических нарушений: избыточное накопление гликогена и липидов, дегенеративные изменения митохондрий только в единичных волокнах, обнаружение скопления большого числа мелких темных митохондрий или появление митохондрий неправильной формы с включением гликогена, таким образом, в мышечной ткани преобладают неспецифические миопатические изменения, морфологические признаки митохондриальной миопатии выявлены только в единичных волокнах.
Со слов матери при МРТ головного мозга выявлены очаги демиелинизации, был поставлен диагноз — рассеянный энцефаломиелит, проводились курсы плазмофереза и гормонотерапии, без эффекта. Пациентка лечилась в США, где клинико-морфологическая картина также была расценена как демиелинизирующий процесс. В 20 лет развился пароксизм с выраженными длительными и генерализующимися миоклониями. Проведено генетическое обследование, биопсия бедренной мышцы, при которой выявлены: Атрофия отдельных мышечных волокон, преимущественно 11 типа, изменение нервно-мышечных пластинок с исчезновением аксонального их компонента, при реакции на SDH только в единичных мышечных волокнах выявляется периферическая зона усиления реакции, свидетельствующая о скоплении митохондрий по периферии мышечного волокна, выявлены признаки миопатических изменений в виде нарушения архитектуры и очагового лизиса миофибриллярного аппарата, в части волокон — признаки метаболических нарушений: избыточное накопление гликогена и липидов, дегенеративные изменения митохондрий только в единичных волокнах, обнаружение скопления большого числа мелких темных митохондрий или появление митохондрий неправильной формы с включением гликогена, таким образом, в мышечной ткани преобладают неспецифические миопатические изменения, морфологические признаки митохондриальной миопатии выявлены только в единичных волокнах. Установлен диагноз: «Митохондриальная энцефалопатия. Синдром MERRF». Родители здоровы, генетически не обследованы. Периодически проходит курсы метаболической и антиоксидантной терапии. Постоянно принимает Коэнзим Q10, Депакин хроно 1500 мг в сут, Фенобарбитал 100 мг на ночь, периодически (во время приступов) Диазепам 5 мг. В течение последней недели появились миоклонические подёргивания мимической мускулатуры справа, речь стала более смазанной. Госпитализирована в экстренном порядке для обследования и лечения. Состояние при поступлении: Общее состояние относительно удовлетворительное. Кожные покровы чистые, бледные. Отеков нет. Над легкими везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. АД 110/70 мм рт. ст. Пульс 68 в мин. Живот при пальпации мягкий, безболезненный во всех отделах. Дизурии нет. Неврологический статус: Сознание ясное, ориентирована полностью. Мнестико-интелектуально снижена. Демонстративна. Речь тихая слабомодулированная. Снижение долговременной памяти.
Установлен диагноз: «Митохондриальная энцефалопатия. Синдром MERRF». Родители здоровы, генетически не обследованы. Периодически проходит курсы метаболической и антиоксидантной терапии. Постоянно принимает Коэнзим Q10, Депакин хроно 1500 мг в сут, Фенобарбитал 100 мг на ночь, периодически (во время приступов) Диазепам 5 мг. В течение последней недели появились миоклонические подёргивания мимической мускулатуры справа, речь стала более смазанной. Госпитализирована в экстренном порядке для обследования и лечения. Состояние при поступлении: Общее состояние относительно удовлетворительное. Кожные покровы чистые, бледные. Отеков нет. Над легкими везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. АД 110/70 мм рт. ст. Пульс 68 в мин. Живот при пальпации мягкий, безболезненный во всех отделах. Дизурии нет. Неврологический статус: Сознание ясное, ориентирована полностью. Мнестико-интелектуально снижена. Демонстративна. Речь тихая слабомодулированная. Снижение долговременной памяти. Менингеальных знаков нет. Поля зрения ориентировочно не сужены. Зрачки D=S. Фотореакции живые, конвергенция ослаблена. Движения глазных яблок не ограничены. Нистагм установочный мелкоразмашистый в крайних отведениях. Миоклонические подёргивания мимической мускулатуры справа. Глоточные рефлексы средней живости. Язык по средней линии. Парезов нет. Гипотония мышц конечностей. Диффузная мышечная гипотрофия. Сухожильные рефлексы живые, S=D. Патологические рефлексы не вызываются. Расстройств чувствительности нет. В позе Ромберга устойчива. Координаторные пробы выполняет удовлетворительно с 2 сторон. Дополнительное обследование: В общем и биохимическом анализе крови без патологических изменений. За исключением Лактата — 3,0. ЭКГ: Ритм синусовый. ЧСС 63 в мин. Отклонение ЭОС вправо. ЭЭГ: Диффузные изименения ЭЭГ, умеренно выраженная дизритмия, за счет неравномерности и недостаточной выраженности основного ритма покоя в сочетании с негрубым замедлением биопотенциалов мозга. В ходе записи регистрируется негрубая неспецифическая островолновая активность с преобладанием над лобно-височными отделами справа, несколько нарастающая на фоне гипервентиляции.
Менингеальных знаков нет. Поля зрения ориентировочно не сужены. Зрачки D=S. Фотореакции живые, конвергенция ослаблена. Движения глазных яблок не ограничены. Нистагм установочный мелкоразмашистый в крайних отведениях. Миоклонические подёргивания мимической мускулатуры справа. Глоточные рефлексы средней живости. Язык по средней линии. Парезов нет. Гипотония мышц конечностей. Диффузная мышечная гипотрофия. Сухожильные рефлексы живые, S=D. Патологические рефлексы не вызываются. Расстройств чувствительности нет. В позе Ромберга устойчива. Координаторные пробы выполняет удовлетворительно с 2 сторон. Дополнительное обследование: В общем и биохимическом анализе крови без патологических изменений. За исключением Лактата — 3,0. ЭКГ: Ритм синусовый. ЧСС 63 в мин. Отклонение ЭОС вправо. ЭЭГ: Диффузные изименения ЭЭГ, умеренно выраженная дизритмия, за счет неравномерности и недостаточной выраженности основного ритма покоя в сочетании с негрубым замедлением биопотенциалов мозга. В ходе записи регистрируется негрубая неспецифическая островолновая активность с преобладанием над лобно-височными отделами справа, несколько нарастающая на фоне гипервентиляции. Типичной эпиактивности на момент исследования не зафиксировано. МРТ головного мозга: На Т1 и Т2 взвешенных изображениях в сагиттальной и аксиаль¬ной проекциях исследования кора и белое вещество головного мозга развиты правильно и имеют нормальную интенсивность мр-сигнала. В базальных ядрах, внутренней капсуле и мозолистом теле не определя¬ется изменений мр-сигнала. Очаговых изменений мр сигнала в стволе и мозжечке не выявлено. Срединные структуры головного мозга не смещены. Желудочки мозга расширены, форма их не изменена. Боковые желудочки симметричны. Выявляется расширение субарахноидального пространства и бо¬розд большого мозга и мозжечка. Турецкое седло и гипофиз не измене¬ны. Параселлярные структуры без особенностей. Дополнительных образований в области мостомозжечковых уг¬лов не выявлено. Внутренние слуховые проходы не расширены, сим¬метричны. Околоносовые пазухи и ячейки сосцевидных отростков височных костей развиты правильно, их пневматизация не нарушена. Глазницы без особенностей.
Типичной эпиактивности на момент исследования не зафиксировано. МРТ головного мозга: На Т1 и Т2 взвешенных изображениях в сагиттальной и аксиаль¬ной проекциях исследования кора и белое вещество головного мозга развиты правильно и имеют нормальную интенсивность мр-сигнала. В базальных ядрах, внутренней капсуле и мозолистом теле не определя¬ется изменений мр-сигнала. Очаговых изменений мр сигнала в стволе и мозжечке не выявлено. Срединные структуры головного мозга не смещены. Желудочки мозга расширены, форма их не изменена. Боковые желудочки симметричны. Выявляется расширение субарахноидального пространства и бо¬розд большого мозга и мозжечка. Турецкое седло и гипофиз не измене¬ны. Параселлярные структуры без особенностей. Дополнительных образований в области мостомозжечковых уг¬лов не выявлено. Внутренние слуховые проходы не расширены, сим¬метричны. Околоносовые пазухи и ячейки сосцевидных отростков височных костей развиты правильно, их пневматизация не нарушена. Глазницы без особенностей.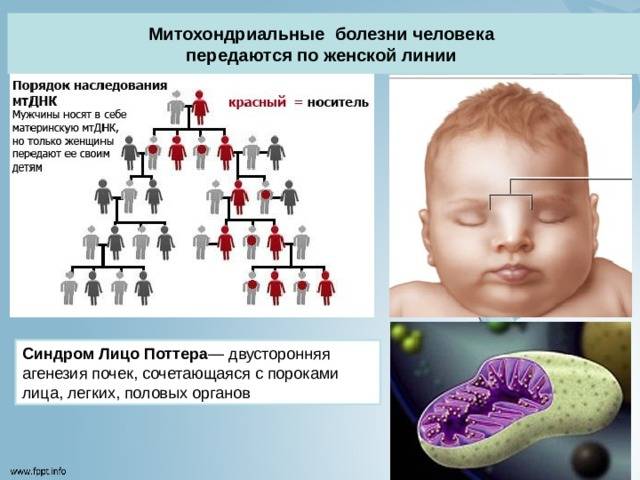 Обращает на себя внимание выраженный гиперостоз лобной и теменных костей. Заключение: Смешанная гидроцефалия. Таким образом у пациентки на 7 год проявления заболевания диагностирована – Митохондриальная энцефалопатия. Синдром MERRF. Данное заболевание диагностируется крайне редко, что вероятнее всего связано с малой информированностью неврологов и трудностями диагностики (генетическое исследование, биопсия мышц, анализ крови на лактат-пируват). Существуют разработанные профессором Научного центра неврологии РАМН С. Н. Иллариошкиным рекомендации по алгоритму диагностики митохондриальных энцефаломиопатий (см. информацию на сайте).
Обращает на себя внимание выраженный гиперостоз лобной и теменных костей. Заключение: Смешанная гидроцефалия. Таким образом у пациентки на 7 год проявления заболевания диагностирована – Митохондриальная энцефалопатия. Синдром MERRF. Данное заболевание диагностируется крайне редко, что вероятнее всего связано с малой информированностью неврологов и трудностями диагностики (генетическое исследование, биопсия мышц, анализ крови на лактат-пируват). Существуют разработанные профессором Научного центра неврологии РАМН С. Н. Иллариошкиным рекомендации по алгоритму диагностики митохондриальных энцефаломиопатий (см. информацию на сайте).
Фотогалерея статьи
4 мая 2016 г.
Ещё больше полезной информации на нашем Telegram канале
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды: MedlinePlus Genetics
Описание
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) — это состояние, поражающее многие системы организма, особенно мозг и нервную систему. системы (энцефало-) и мышц (миопатия). Признаки и симптомы этого расстройства чаще всего появляются в детстве после периода нормального развития, хотя они могут начаться в любом возрасте. Ранние симптомы могут включать мышечную слабость и боль, повторяющиеся головные боли, потерю аппетита, рвоту и судороги. У большинства больных наблюдаются эпизоды, подобные инсульту, которые начинаются до 40 лет. Эти эпизоды часто включают временную мышечную слабость на одной стороне тела (гемипарез), изменение сознания, нарушения зрения, судороги и сильные головные боли, напоминающие мигрень. Повторяющиеся эпизоды, подобные инсульту, могут постепенно повреждать мозг, приводя к потере зрения, проблемам с движением и потере интеллектуальной функции (слабоумие).
системы (энцефало-) и мышц (миопатия). Признаки и симптомы этого расстройства чаще всего появляются в детстве после периода нормального развития, хотя они могут начаться в любом возрасте. Ранние симптомы могут включать мышечную слабость и боль, повторяющиеся головные боли, потерю аппетита, рвоту и судороги. У большинства больных наблюдаются эпизоды, подобные инсульту, которые начинаются до 40 лет. Эти эпизоды часто включают временную мышечную слабость на одной стороне тела (гемипарез), изменение сознания, нарушения зрения, судороги и сильные головные боли, напоминающие мигрень. Повторяющиеся эпизоды, подобные инсульту, могут постепенно повреждать мозг, приводя к потере зрения, проблемам с движением и потере интеллектуальной функции (слабоумие).
У большинства людей с MELAS в организме накапливается молочная кислота, что называется лактоацидозом. Повышенная кислотность крови может привести к рвоте, болям в животе, крайней усталости (усталости), мышечной слабости и затрудненному дыханию. Реже люди с MELAS могут испытывать непроизвольные мышечные спазмы (миоклонус), нарушение координации мышц (атаксия), потерю слуха, проблемы с сердцем и почками, диабет и гормональный дисбаланс.
Реже люди с MELAS могут испытывать непроизвольные мышечные спазмы (миоклонус), нарушение координации мышц (атаксия), потерю слуха, проблемы с сердцем и почками, диабет и гормональный дисбаланс.
Частота
Точная заболеваемость MELAS неизвестна. Это одно из наиболее распространенных состояний в группе, известной как митохондриальные заболевания. В совокупности митохондриальные заболевания встречаются примерно у 1 из 4000 человек.
Причины
MELAS может быть результатом мутаций в одном из нескольких генов, включая MT-ND1 , MT-ND5 , MT-TH , MT-TL1 и MT- ТВ . Эти гены находятся в ДНК клеточных структур, называемых митохондриями, которые преобразуют энергию из пищи в форму, которую могут использовать клетки. Хотя большая часть ДНК упакована в хромосомы внутри ядра, митохондрии также имеют небольшое количество собственной ДНК, известной как митохондриальная ДНК или мтДНК.
Некоторые гены, связанные с MELAS, предоставляют инструкции для создания белков, участвующих в нормальной функции митохондрий. Эти белки являются частью большого комплекса ферментов в митохондриях, которые помогают преобразовывать кислород, жиры и простые сахара в энергию. Другие гены, связанные с этим заболеванием, предоставляют инструкции для создания молекул, называемых транспортными РНК (тРНК), которые являются химическими родственниками ДНК. Эти молекулы помогают собирать белковые строительные блоки, называемые аминокислотами, в полноразмерные функционирующие белки в митохондриях.
Эти белки являются частью большого комплекса ферментов в митохондриях, которые помогают преобразовывать кислород, жиры и простые сахара в энергию. Другие гены, связанные с этим заболеванием, предоставляют инструкции для создания молекул, называемых транспортными РНК (тРНК), которые являются химическими родственниками ДНК. Эти молекулы помогают собирать белковые строительные блоки, называемые аминокислотами, в полноразмерные функционирующие белки в митохондриях.
Мутации в конкретном гене транспортной РНК, MT-TL1 , вызывают более 80 процентов всех случаев MELAS. Эти мутации нарушают способность митохондрий производить белки, использовать кислород и производить энергию. Исследователи не определили, как изменения в мтДНК приводят к конкретным признакам и симптомам MELAS. Они продолжают исследовать эффекты мутаций митохондриальных генов в различных тканях, особенно в головном мозге.
Наследование
Это состояние наследуется по митохондриальному типу, который также известен как наследование по материнской линии. Этот тип наследования относится к генам, содержащимся в мтДНК. Поскольку яйцеклетки, а не сперматозоиды, вносят митохондрии в развивающийся эмбрион, дети могут наследовать только нарушения, возникающие в результате мутаций мтДНК, от матери. Эти расстройства могут проявляться в каждом поколении семьи и затрагивать как мужчин, так и женщин, но отцы не передают своим детям признаки, связанные с изменениями мтДНК.
Этот тип наследования относится к генам, содержащимся в мтДНК. Поскольку яйцеклетки, а не сперматозоиды, вносят митохондрии в развивающийся эмбрион, дети могут наследовать только нарушения, возникающие в результате мутаций мтДНК, от матери. Эти расстройства могут проявляться в каждом поколении семьи и затрагивать как мужчин, так и женщин, но отцы не передают своим детям признаки, связанные с изменениями мтДНК.
В большинстве случаев люди с MELAS наследуют измененный митохондриальный ген от своей матери. Реже заболевание возникает в результате новой мутации митохондриального гена и возникает у людей без семейного анамнеза MELAS.
Другие названия этого состояния
- MELAS
- MELAS-синдром
- Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды
- Митохондриальная миопатия, лактоацидоз, инсультоподобный эпизод
- Миопатия, митохондриальная-энцефалопатия-молочнокислый ацидоз-инсульт
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: ювенильная миопатия, энцефалопатия, лактоацидоз И инсульт
Информационный центр генетических и редких заболеваний
- Митохондриальная энцефаломиопатия лактоацидоз и инсультоподобные эпизоды
Ресурсы поддержки пациентов и защиты интересов
- Информационный поиск по болезни
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov
- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, ЭНЦЕФАЛОПАТИЯ, ЛАКТАЦИДОЗ И ИНСУЛЬТОПОДОБНЫЕ ЭПИЗОДЫ
Научные статьи в PubMed
- PubMed
Ссылки
- Беттс Дж., Ярос Э., Перри Р.Х., Шефер А.М., Тейлор Р.В., Абдель-Алл З., Лайтоулерс
РН, Тернбулл Д.М. Молекулярная нейропатология MELAS: уровень гетероплазмии в
отдельные нейроны и признаки обширного вовлечения сосудов. невропатол
Заявл. Нейробиол. 2006 авг; 32 (4): 359-73. doi: 10.1111/j.1365-2990.2006.00731.x.
Цитата в PubMed - Эль-Хаттаб А.В., Альманнаи М., Скалья Ф. МЕЛАС. 27 февраля 2001 г. [обновлено 29 ноября 2018 г.].
Пришли: Адам М.П., Мирзаа Г.М., Пагон Р.А., Уоллес С.Е., Бин Л.Дж.Х., Грипп К.В., Амемия А.,
редакторы. GeneReviews(R) [Интернет]. Сиэтл (Вашингтон): Университет
Вашингтон, Сиэтл; 1993-2023. Доступна с
http://www.ncbi.nlm.nih. gov/books/NBK1233/
gov/books/NBK1233/
Цитата в PubMed - Гудфеллоу Дж. А., Дэни К., Стюарт В., Сантош С., Маклин Дж., Малхерн С., Разви С.
Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды:
важной причиной инсульта у молодых людей. Postgrad Med J. 2012
Июнь; 88 (1040): 326-34. doi: 10.1136/postgradmedj-2011-130326. Epub 2012, 10 февраля.
Цитата в PubMed - Мацумото Дж., Сэвер Дж.Л., Бреннан К.С., Рингман Дж.М. Митохондриальная энцефаломиопатия
с лактоацидозом и инсультом (MELAS). Преподобный Нейрол Дис. 2005 Зима; 2 (1): 30-4.
Цитата на PubMed - Sproule DM, Kaufmann P. Митохондриальная энцефалопатия, лактоацидоз и
инсультоподобные эпизоды: основные понятия, клинический фенотип и терапевтические
Лечение синдрома MELAS. Энн Н.Ю. Академия наук. 2008 Октябрь; 1142: 133-58. дои:
10.1196/летопись.1444.011. Erratum In: Ann NY Acad Sci. 2009 Апрель; 1161: 601. Цитата в PubMed - Тамбисетти М.
 , Ньюман, штат Нью-Джерси. Диагностика и лечение MELAS. Эксперт Рев Мол
, Ньюман, штат Нью-Джерси. Диагностика и лечение MELAS. Эксперт Рев Мол
Диагн. 2004 г., сен; 4(5):631-44. дои: 10.1586/14737159.4.5.631. Цитата в PubMed
Синдром Меласа — StatPearls — NCBI Bookshelf
Непрерывное обучение
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) — это митохондриальное заболевание, поражающее преимущественно нервную систему и мышцы. MELAS проявляется у детей или молодых людей в виде повторяющихся эпизодов энцефалопатии, миопатии, головной боли и очагового неврологического дефицита. Состояние неуклонно прогрессирует, приводя к неврологическим нарушениям в подростковом или раннем взрослом возрасте. Это упражнение описывает патофизиологию и проявления синдрома MELAS и подчеркивает роль межпрофессиональной команды в его лечении.
Цели:
Рассмотрите причину синдрома MELAS.
Опишите проявления синдрома MELAS.
Обобщите дифференциальную диагностику синдрома MELAS.

Объясните важность улучшения координации помощи между членами межпрофессиональной бригады для улучшения результатов лечения пациентов с синдромом MELAS.
Получите доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Введение
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) — это митохондриальное заболевание , в первую очередь поражающее нервную систему и мышцы. MELAS проявляется у детей или молодых людей в виде повторяющихся эпизодов энцефалопатии, миопатии, головной боли и очагового неврологического дефицита. Состояние неуклонно прогрессирует, приводя к неврологическим нарушениям в подростковом или раннем взрослом возрасте.
Этиология
MELAS — это митохондриальное наследуемое генетическое заболевание. Отцовские митохондрии присутствуют только в хвостовой части сперматозоидов. В результате они теряются во время оплодотворения, а митохондриальные нарушения, включая MELAS, наследуются по материнской линии. В редких случаях MELAS может быть результатом спорадической мутации без семейного анамнеза. Митохондриальные генетические нарушения являются результатом мутаций, вызывающих нарушение функции митохондрий, включая окислительное фосфорилирование и выработку энергии. Считается, что при MELAS мутации тРНК вызывают нарушение сборки белков в комплексы дыхательной цепи, хотя точные механизмы еще предстоит выяснить. Митохондрии являются электростанцией клеток. Любое митохондриальное расстройство влияет на наиболее метаболически активные органы тела, особенно на мозг и мышцы.
В редких случаях MELAS может быть результатом спорадической мутации без семейного анамнеза. Митохондриальные генетические нарушения являются результатом мутаций, вызывающих нарушение функции митохондрий, включая окислительное фосфорилирование и выработку энергии. Считается, что при MELAS мутации тРНК вызывают нарушение сборки белков в комплексы дыхательной цепи, хотя точные механизмы еще предстоит выяснить. Митохондрии являются электростанцией клеток. Любое митохондриальное расстройство влияет на наиболее метаболически активные органы тела, особенно на мозг и мышцы.
Многие различные мутации транспортной РНК (тРНК) могут вызывать MELAS. Наиболее распространена мутация в митохондриальном гене MTTL1. Мутация с одной парой оснований, m.3243A>G, обнаруживается у 80% пациентов, а вторая распространенная мутация, m.3271T>C, обнаруживается у 10%.[1][2] Однако многие другие гены идентифицируются со сходными фенотипическими синдромами, такими как POLG и BCS1L.
Считается, что неврологические симптомы MELAS возникают в результате сочетания нарушения выработки митохондриальной энергии, микрососудистой ангиопатии и дефицита оксида азота, что может вызвать нарушение церебральной вазодилатации. [3]
[3]
Эпидемиология
MELAS является одним из наиболее распространенных митохондриальных заболеваний, частота возникновения которого оценивается как 1 случай на 4000.
Оба пола страдают в равной степени, но только женщины могут передавать заболевание, поскольку митохондрии переносятся в хвостах сперматозоидов. и, следовательно, выходят за пределы зиготы во время оплодотворения.
Патофизиология
Каждая соматическая клетка обладает множественными и изменчивыми копиями одной и той же митохондриальной ДНК или генома. Эта митохондриальная ДНК также повреждается в разной степени при старении клеток. Как и любое митохондриальное заболевание, MELAS проявляет гетероплазмию или вариации типов митохондриальной ДНК, обнаруженных в разных тканях одного и того же человека. Обычно генетические мутации затрагивают только некоторые митохондрии, что приводит к различному появлению больных митохондрий в разных тканях у одного и того же человека. Это явление объясняет широкую вариабельность проявления различных тканей у одного человека, а также проявления у разных пациентов с одним и тем же диагнозом. Это также имеет значение для диагностики, поскольку иногда исследования сыворотки и мочи могут быть отрицательными, если эти клеточные линии не затронуты, что делает необходимым выполнение биопсии мышц для исследования пораженной ткани.
Это также имеет значение для диагностики, поскольку иногда исследования сыворотки и мочи могут быть отрицательными, если эти клеточные линии не затронуты, что делает необходимым выполнение биопсии мышц для исследования пораженной ткани.
Митохондриальные заболевания, включая MELAS, поражают в основном ткани с наивысшей метаболической активностью. К ним относятся скелетные и сердечные мышцы, головной мозг, глаза и внутреннее ухо. Из-за нарушения функции митохондриальной дыхательной цепи и окислительного фосфорилирования повышенная анаэробная гликолитическая активность приводит к увеличению молочной кислоты во время острых приступов.
Гистопатология
Отличительным признаком митохондриального заболевания является скопление митохондрий в мышечных волокнах, визуализируемое при окрашивании трихромом по Гомори. Больные митохондрии размножаются в попытке компенсировать плохое производство энергии и кажутся ярко-красными по сравнению с синими миофибриллами, поэтому их называют «рваными красными волокнами». MELAS также показывает сильно реактивные сукцинатдегидрогеназу (SDH) кровеносные сосуды из-за митохондриальной пролиферации в периваскулярных гладкомышечных и эндотелиальных клетках.
MELAS также показывает сильно реактивные сукцинатдегидрогеназу (SDH) кровеносные сосуды из-за митохондриальной пролиферации в периваскулярных гладкомышечных и эндотелиальных клетках.
Анамнез и физикальное исследование
Дети с MELAS часто имеют нормальное раннее психомоторное развитие до появления симптомов в возрасте от 2 до 10 лет. Хотя и реже, но может иметь место младенческое начало, которое может проявляться задержкой развития, задержкой роста и прогрессирующей глухотой. Начало у детей старшего возраста обычно проявляется повторяющимися приступами мигренеподобной головной боли, анорексии, рвоты и судорог. Дети с MELAS также часто имеют низкий рост.
Миопатия при MELAS первоначально проявляется непереносимостью физической нагрузки или слабостью проксимальных мышц. Из-за тяжести других неврологических особенностей миопатия часто не распознается. Миопатия при MELAS часто не вызывает значительных функциональных нарушений.
Приступы при MELAS могут быть фокальными или генерализованными и не имеют признанной симптоматики, специфичной для синдрома, или результатов ЭЭГ. Дети с более ранним возрастом, как правило, имеют более высокие показатели лекарственно-резистентной эпилепсии и, следовательно, более тяжелую клиническую дисфункцию.
Дети с более ранним возрастом, как правило, имеют более высокие показатели лекарственно-резистентной эпилепсии и, следовательно, более тяжелую клиническую дисфункцию.
Судороги часто сопровождаются внезапными очаговыми неврологическими нарушениями, которые называются «инсультоподобными эпизодами». Эти эпизоды отличаются от типичного инсульта тем, что они не соответствуют какой-либо сосудистой территории, результаты МРТ могут колебаться или разрешаться быстрее, чем при типичном инсульте, а кажущийся коэффициент диффузии на МРТ не всегда снижен, как при типичном инфаркте, а вместо этого может быть повышенный или смешанный. Типичные очаговые неврологические расстройства включают гемипарез, гемианопсию или афазию. Остаточные дефициты после инсультоподобных эпизодов постепенно нарушают неврологическую функцию. К подростковому или юношескому взрослому возрасту у пациентов часто возникают нарушения двигательной функции, зрения, мышления и нейросенсорная тугоухость.
Энцефалопатия — еще одна ключевая особенность. Измененный уровень сознания часто связан с инсультоподобными эпизодами. Психическое ухудшение начинается в детстве и медленно прогрессирует.
Измененный уровень сознания часто связан с инсультоподобными эпизодами. Психическое ухудшение начинается в детстве и медленно прогрессирует.
В MELAS могут быть вовлечены и другие системы органов. Сообщается, что у пациентов отмечаются нарушения сердечной проводимости, сахарный диабет и хроническая усталость.
Оценка
МРТ: мультифокальные инфарктоподобные области коры головного мозга на разных стадиях ишемической эволюции, области, не соответствующие ни одной известной сосудистой территории. Начальные поражения часто возникают в затылочных или теменных долях с последующим поражением мозжечка, коры головного мозга, базальных ганглиев и таламуса.
Лактат часто повышен в сыворотке и спинномозговой жидкости (ЦСЖ). МР-спектроскопия может показать повышенный пик лактата в пораженных и даже непораженных областях мозга.
Биопсия мышц показывает рваные красные волокна. Однако сначала следует провести генетическую оценку, что в большинстве случаев устраняет необходимость в биопсии мышц.
Диагноз может быть молекулярным или клиническим:
Инсультоподобные эпизоды в возрасте до 40 лет
Энцефалопатия с судорогами или деменцией
Лактоацидоз в крови* или рваные красные волокна при биопсии мышц
остановить прогрессирование болезни.
Симптоматическое лечение судорог противоэпилептическими препаратами. Есть несколько сообщений об обострении эпилепсии MELAS при применении вальпроата, хотя механизмы этого взаимодействия все еще изучаются.
Считается, что витамины, такие как коэнзим Q10 или L-карнитин, помогают увеличить выработку энергии митохондриями и могут замедлить последствия болезни. В настоящее время проводятся испытания фазы I и II MELAS идебенона, синтетического кофермента Q10, который, как было показано, улучшает неврологическую функцию при других митохондриальных заболеваниях (Scaglia, ClinicalTrials.gov, идентификатор: NCT00887562)
Было показано, что L-аргинин ослабляет тяжесть симптомов при применении в острых приступах и уменьшение частоты эпизодов[4][3]. Также считается, что L-цитруллин полезен для восстановления и снижения риска инсульта. Предполагается, что эта взаимосвязь связана с коррекцией дефицита оксида азота у пациентов с MELAS, поскольку аргинин и цитруллин являются предшественниками образования оксида азота.
Также считается, что L-цитруллин полезен для восстановления и снижения риска инсульта. Предполагается, что эта взаимосвязь связана с коррекцией дефицита оксида азота у пациентов с MELAS, поскольку аргинин и цитруллин являются предшественниками образования оксида азота.
Дифференциальный диагноз
Другие митохондриальные нарушения следует рассматривать у пациентов с семейным анамнезом, касающимся материнского митохондриального наследования.
Кернс-Сейр — редкое нервно-мышечное заболевание, которое также может проявляться нарушением зрения, низким ростом, потерей слуха и атаксией. Однако Kearns-Sayre отличается характерными визуальными находками:
Хроническая прогрессирующая наружная офтальмоплегия
Пигментный атипичный ретинит
Пигментная дегенерация сетчатки
В дополнение к этим визуальным симптомам у пациентов с болезнью Кернс-Сейр часто наблюдаются сердечные проявления, которые помогают отличить этих пациентов от пациентов с MELAS.
Миоклонус-эпилепсия, связанная с рваными красными волокнами (MERRF), может быть перепутана с MELAS, так как они оба включают судороги, ухудшение психического состояния и миопатию с рваными красными волокнами при биопсии. Пациенты с MERRF также могут иметь потерю слуха, нарушения зрения, вторичные по отношению к атрофии зрительного нерва, и низкий рост. Характерные миоклонические судороги при MERRF могут помочь сузить диагностику, но следует рассмотреть генетическое тестирование, чтобы различить эти два состояния.
Синдром Лея также может проявляться прогрессирующим неврологическим ухудшением, судорогами и рвотой, главным образом у маленьких детей.
Повышение эффективности медицинских работников
Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) — это митохондриальное заболевание , поражающее преимущественно нервную систему и мышцы. MELAS проявляется у детей или молодых людей в виде повторяющихся эпизодов энцефалопатии, миопатии, головной боли и очагового неврологического дефицита. Поскольку это состояние неуклонно прогрессирует, приводя к неврологическим нарушениям в подростковом или раннем взрослом возрасте, лучше всего с ним справляется межпрофессиональная команда. У большинства пациентов лечение является поддерживающим, но общий прогноз неблагоприятный. Важно привлечь социального работника и физиотерапевта на ранних стадиях заболевания, потому что эти специалисты могут помочь улучшить качество жизни.[5][3][6][7][8]
Поскольку это состояние неуклонно прогрессирует, приводя к неврологическим нарушениям в подростковом или раннем взрослом возрасте, лучше всего с ним справляется межпрофессиональная команда. У большинства пациентов лечение является поддерживающим, но общий прогноз неблагоприятный. Важно привлечь социального работника и физиотерапевта на ранних стадиях заболевания, потому что эти специалисты могут помочь улучшить качество жизни.[5][3][6][7][8]
Контрольные вопросы
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Ссылки
- 1.
Икеда Т., Осака Х., Шимбо Х., Таджика М., Ямадзаки М., Уэда А., Мураяма К., Ямагата Т. Мутация митохондриальной ДНК 3243A>T в пациент с синдромом MELAS. Гум Геном Вар. 2018;5:25. [Бесплатная статья PMC: PMC6123423] [PubMed: 30210801]
- 2.
Niedermayr K, Pölzl G, Scholl-Bürgi S, Fauth C, Schweigmann U, Haberlandt E, Albrecht U, Zlamy M, Sperl W, Mayr JA, Karall D.
 Мутация митохондриальной ДНК «m.3243A>G» — гетерогенная клиническая картинка для кардиологов («m.3243A>G»: Фенотипический хамелеон). Врожденный порок сердца. 2018 сен; 13 (5): 671-677. [PubMed: 30133155]
Мутация митохондриальной ДНК «m.3243A>G» — гетерогенная клиническая картинка для кардиологов («m.3243A>G»: Фенотипический хамелеон). Врожденный порок сердца. 2018 сен; 13 (5): 671-677. [PubMed: 30133155]- 3.
Koga Y, Povalko N, Inoue E, Nakamura H, Ishii A, Suzuki Y, Yoneda M, Kanda F, Kubota M, Okada H, Fujii K. Терапевтический режим L -аргинин для МЕЛАС: 9-летнее, проспективное, многоцентровое, клиническое исследование. Дж Нейрол. 2018 декабрь; 265(12):2861-2874. [Бесплатная статья PMC: PMC6244654] [PubMed: 30269300]
- 4.
Radelfahr F, Klopstock T. [Диагностические и терапевтические подходы к митохондриальным заболеваниям]. Fortschr Neurol Psychiatr. 2018 сен; 86 (9): 584-591. [PubMed: 30248691]
- 5.
Брамбилла А., Фавилли С., Оливотто И., Калабри Г.Б., Порседа Г., Де Симоне Л., Прокопио Э., Пасквини Э., Донати М.А. Клинический профиль и исход поражения сердца при синдроме MELAS. Int J Кардиол. 201901 фев; 276:14-19.
