Метаболический кетоацидоз у детей: Ацетонемический синдром

КЕТОАЦИДОЗ И КЕТОАЦИДОТИЧЕСКАЯ КОМА | Демидова И.Ю.
Диагностика диабетического кетоацидоза при установленном сахарном диабете не вызывает затруднений. Особого внимания требуют случаи, когда сахарный диабет манифестирует в состоянии кетоацидоза. Представлены рекомендации по лечению этого состояния и его осложнений.
To diagnose diabetic ketoacidosis in documented diabetes mellitus presents no difficulties. Emphasis should be laid on the cases in which diabetes mellitus is manifestative in the presence of ketoacidosis. Recommendations for treatment of this condition and its complications are given.
И.Ю. Демидова — кафедра эндокринологии ММА им. И.М. Сеченова (зав. — акад. РАМН проф. И.И. Дедов)
I.Yu. Demidova — Department of Endocrinology (Head Prof. I.I.Dedov, Academician of the Russian Academy of Medical Sciences, I.M.Sechenov Moscow Medical Academy
Кетоацидоз и кетоацидотическая кома являются одной из основных причин смерти больных сахарным диабетом (СД) в возрасте до 20 лет. Более 16% пациентов, страдающих инсулинзависимым СД (ИЗСД), умирают именно от кетоацидоза или кетоацидотической комы. Риск летального исхода кетоацидоза особенно возрастает в тех случаях, когда фактором, провоцирующим возникновение данного острого осложнения СД, является тяжелое интеркуррентное заболевание.
Более 16% пациентов, страдающих инсулинзависимым СД (ИЗСД), умирают именно от кетоацидоза или кетоацидотической комы. Риск летального исхода кетоацидоза особенно возрастает в тех случаях, когда фактором, провоцирующим возникновение данного острого осложнения СД, является тяжелое интеркуррентное заболевание.
Выявление ИЗСД на ранних стадиях снизило частоту случаев манифестации данного заболевания в состоянии кетоацидоза до 20%. Обучение больных, страдающих СД, принципам самоконтроля и тактике поведения при неотложных состояниях позволило значительно снизить риск возникновения кетоацидоза – до 0, 5-2% случаев в год.
Изучение нюансов патогенеза кетоацидоза и создание оптимальных схем терапии этого состояния привели к снижению частоты летальных исходов, однако смертность от кетоацидотической комы составляет 7 – 19%, а в неспециализированных лечебных учреждениях этот показатель выше.
Патогенез
Наиболее частыми провоцирующими факторами декомпенсации СД и развития кетоацидоза являются любые интеркуррентные заболевания (острые воспалительные процессы, обострения хронических заболеваний, инфекционные болезни), хирургические вмешательства, травмы, нарушения режима лечения (введение просроченного или неправильно хранившегося инсулина, ошибки в назначении или введении дозы препарата, неисправность в системах введения инсулина, эмоциональные стрессовые ситуации, беременность и прекращение введения инсулина с суицидальной целью.
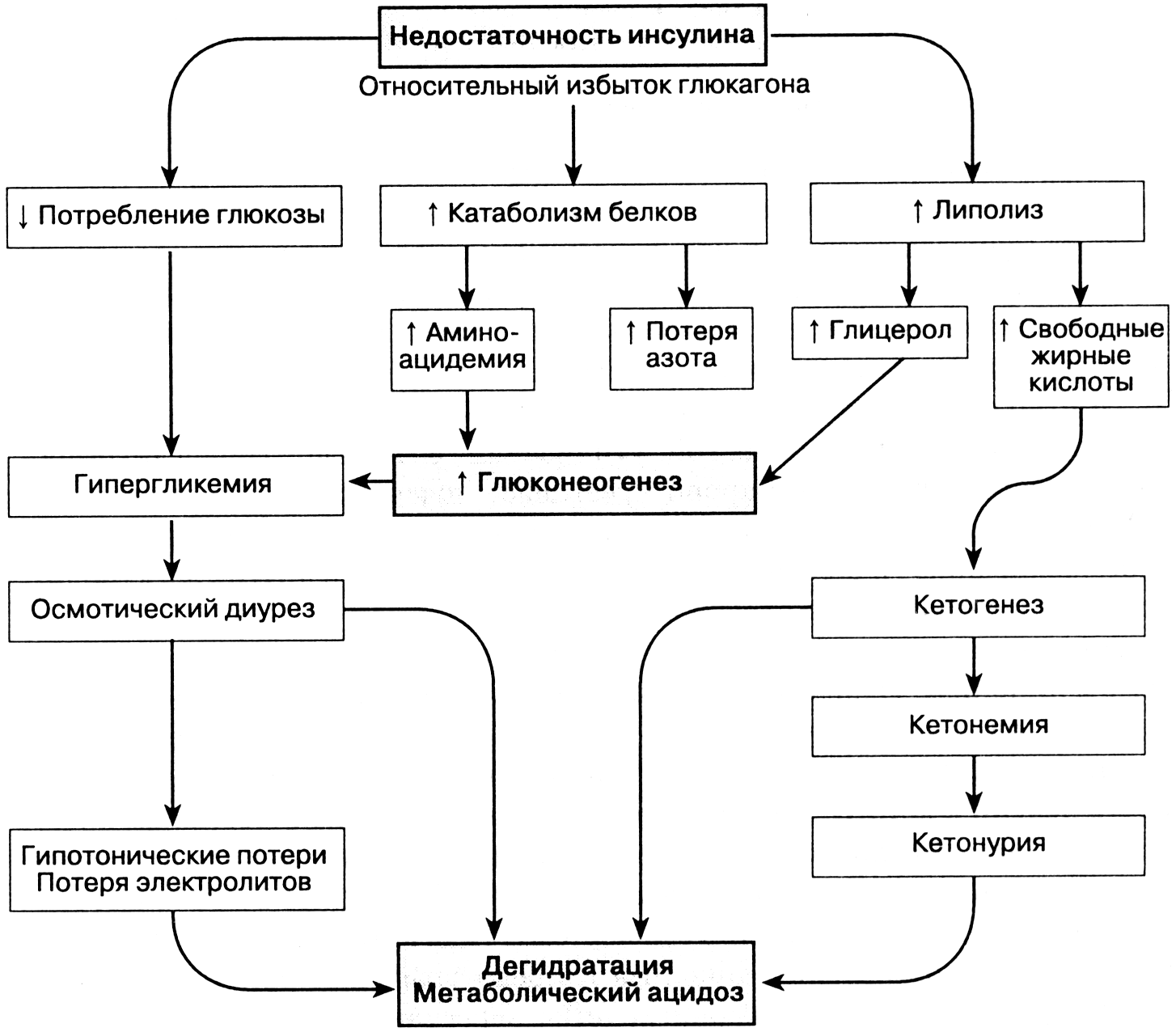
Ведущую роль в патогенезе кетоацидоза играет абсолютная инсулиновая недостаточность, приводящая к снижению утилизации глюкозы инсулинзависимыми тканями и, соответственно гипергликемии, и тяжелому энергетическому голоду в них. Последнее обстоятельство является причиной резкого повышения в крови уровня всех контринсулиновых гормонов (глюкагона, кортизола, катехоламинов, АКТГ, СТГ), стимуляции процессов гликогенолиза, протеолиза и липолиза, поставляющих субстраты для глюконеогенеза в печени и в меньшей степени в почках. Глюконеогенез в сочетании с прямым нарушением утилизации глюкозы тканями в связи с абсолютным дефицитом инсулина является важнейшей причиной быстро нарастающей гипергликемии, повышения осмолярности плазмы, внутриклеточной дегидратации и осмотического диуреза.
Перечисленные факторы приводят к тяжелой внеклеточной дегидратации, гиповолемическому шоку и к значительным электролитным нарушениям. Дегидратация и гиповолемия являются причиной снижения мозгового, почечного и периферического кровотока, что, в свою очередь, усиливает имеющуюся гипоксию ЦНС и периферических тканей и приводит к развитию олигурии и анурии. Гипоксия периферических тканей способствует активизации в них процессов анаэробного гликолиза и постепенному нарастанию уровня лактата. Относительный дефицит лактатдегидрогеназы при дефиците инсулина и невозможность полной утилизации лактата в цикле Кори являются причиной возникновения лактацидоза при декомпенсации ИЗСД. Дефицит инсулина и резкое повышение концентрации всех контринсулиновых гормонов являются причиной активизации липолиза и мобилизации свободных жирных кислот (СЖК), что способствует активной продукции кетоновых тел. Усиленное образование ацетил-КоА, предшественника ацетоацетата (и ацетона при его декарбоксилировании), и В-гидроксибутирата обеспечивается в данных условиях активным поступлением СЖК в печень вследствие их мобилизации из периферических тканей и преобладанием процессов липолиза над липогенезом в самой клетке печени.
Гипоксия периферических тканей способствует активизации в них процессов анаэробного гликолиза и постепенному нарастанию уровня лактата. Относительный дефицит лактатдегидрогеназы при дефиците инсулина и невозможность полной утилизации лактата в цикле Кори являются причиной возникновения лактацидоза при декомпенсации ИЗСД. Дефицит инсулина и резкое повышение концентрации всех контринсулиновых гормонов являются причиной активизации липолиза и мобилизации свободных жирных кислот (СЖК), что способствует активной продукции кетоновых тел. Усиленное образование ацетил-КоА, предшественника ацетоацетата (и ацетона при его декарбоксилировании), и В-гидроксибутирата обеспечивается в данных условиях активным поступлением СЖК в печень вследствие их мобилизации из периферических тканей и преобладанием процессов липолиза над липогенезом в самой клетке печени.
Быстрое нарастание концентрации кетоновых тел при декомпенсации СД обусловлено не только их усиленной продукцией, но и снижением их периферической утилизации и экскреции с мочой в связи с дегидратацией и олигурией, сменившей полиурию. Диссоциация кетоновых тел сопровождается эквимолярной продукцией ионов водорода. В условиях декомпенсации СД продукция кетоновых тел и, следовательно, образование ионов водорода превышают буферную способность тканей и жидкостей организма, что приводит к развитию тяжелого метаболического ацидоза [1].
Диссоциация кетоновых тел сопровождается эквимолярной продукцией ионов водорода. В условиях декомпенсации СД продукция кетоновых тел и, следовательно, образование ионов водорода превышают буферную способность тканей и жидкостей организма, что приводит к развитию тяжелого метаболического ацидоза [1].
Тяжесть состояния при кетоацидозе обусловлена резкой дегидратацией организма, декомпенсированным метаболическим ацидозом, выраженным дефицитом электролитов (калия, натрия, фосфора, магния и др.), гипоксией, гиперосмолярностью (в большинстве случаев) и нередко сопутствующим интеркуррентным заболеванием.
Клиническая картина
Кетоацидоз развивается постепенно в течение нескольких дней. При наличии тяжелой сопутствующей инфекции клиническая картина кетоацидоза разворачивается в более сжатые сроки.
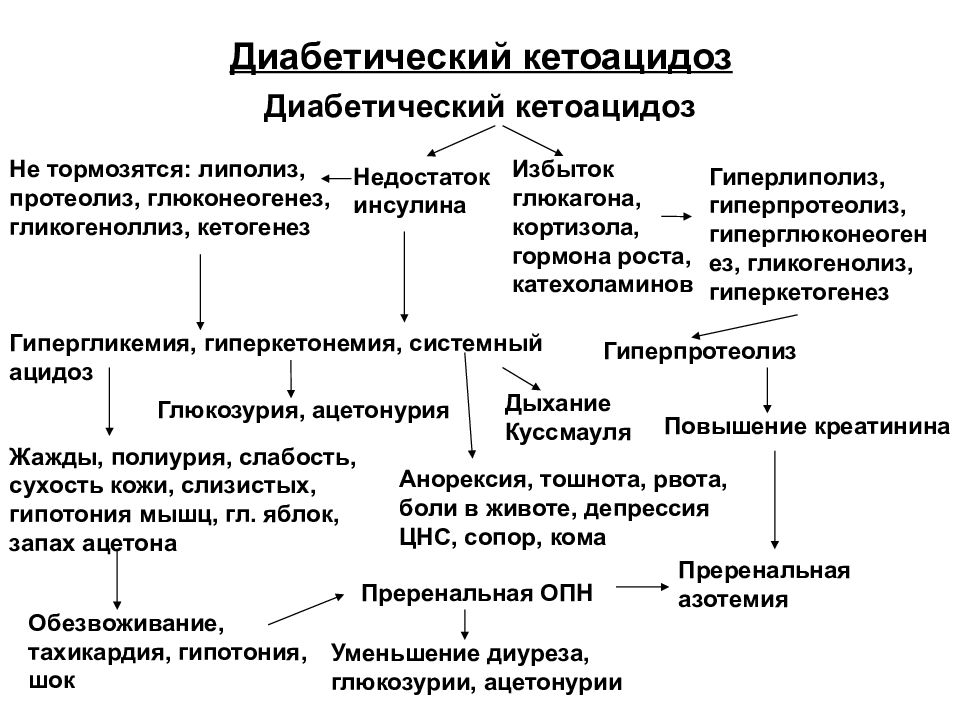
Ранними клиническими симптомами кетоацидоза являются типичные признаки декомпенсации СД, такие как нарастающие сухость слизистых и кожи, жажда, полиурия, впоследствии сменяющаяся олигурией и анурией, слабость, головная боль, сонливость, снижение аппетита, потеря массы тела, появление легкого запаха ацетона в выдыхаемом воздухе. В случае неоказания своевременной помощи метаболические нарушения усугубляются, а описанные выше клинические признаки дополняются неспецифическими симптомами интоксикации и ацидоза, такими как головная боль, головокружение, тошнота и рвота, которая вскоре учащается и приобретает неукротимый характер. Рвотные массы при кетоацидозе нередко имеют кровянисто-коричневатый оттенок и врачи ошибочно принимают это за рвоту “кофейной гущей”. По мере нарастания кетоацидоза запах ацетона в выдыхаемом воздухе усиливается, а дыхание становится частым, шумным и глубоким (респираторная компенсация, дыхание Куссмауля).
В случае неоказания своевременной помощи метаболические нарушения усугубляются, а описанные выше клинические признаки дополняются неспецифическими симптомами интоксикации и ацидоза, такими как головная боль, головокружение, тошнота и рвота, которая вскоре учащается и приобретает неукротимый характер. Рвотные массы при кетоацидозе нередко имеют кровянисто-коричневатый оттенок и врачи ошибочно принимают это за рвоту “кофейной гущей”. По мере нарастания кетоацидоза запах ацетона в выдыхаемом воздухе усиливается, а дыхание становится частым, шумным и глубоким (респираторная компенсация, дыхание Куссмауля).
Заслуживает особого внимания симптом, наблюдаемый более чем у половины больных – так называемый “абдоминальный синдром” кетоацидоза, проявляющийся клиникой “острого живота”. Нередко сочетание болей в животе, рвоты и наблюдаемого при кетоацидозе лейкоцитоза приводит к диагностическим ошибкам и недопустимым в данном состоянии хирургическим вмешательствам, часто заканчивающимся летально. Риск подобных ошибок особенно велик в случае манифестации СД в состоянии кетоацидоза.
Риск подобных ошибок особенно велик в случае манифестации СД в состоянии кетоацидоза.
При объективном осмотре отмечаются выраженные признаки обезвоживания (в тяжелых случаях больные теряют до 10 – 12% массы тела). Тургор тканей резко снижается. Глазные яблоки становятся мягкими, а кожные покровы и видимые слизистые – сухими. Язык обложен густым коричневым налетом. Мышечный тонус, сухожильные рефлексы, температура тела и артериальное давление снижены. Определяется частый пульс слабого наполнения и напряжения. Печень, как правило, значительно выступает из-под края реберной дуги и болезненна при пальпации. Дыхание Куссмауля сопровождается резким запахом ацетона в выдыхаемом воздухе.
При осмотре больных в состоянии кетоацидоза необходимо как можно быстрее уточнить причину, спровоцировавшую декомпенсацию СД. При наличии сопутствующего интеркуррентного заболевания следует немедленно приступить к его лечению.
С первых признаков декомпенсации СД у больных отмечаются признаки сначала легкого, а затем все более и более выраженного угнетения ЦНС. Так, сначала пациенты жалуются на головную боль, становятся раздражительными, а затем – вялыми, апатичными, сонливыми. Развивающееся состояние оглушенности характеризуется снижением уровня бодрствования, замедлением сознательных реакций на раздражители и увеличением периодов сна. По мере усугубления метаболических нарушений состояние оглушенности ступором, часто называемым прекоматозным состоянием, клинически проявляющимся глубоким сном или аналогичной ему по поведенческим реакциям ареактивностью. Конечной стадией нарастающего угнетения ЦНС является кома, характеризующаяся полным отсутствием сознания.
Так, сначала пациенты жалуются на головную боль, становятся раздражительными, а затем – вялыми, апатичными, сонливыми. Развивающееся состояние оглушенности характеризуется снижением уровня бодрствования, замедлением сознательных реакций на раздражители и увеличением периодов сна. По мере усугубления метаболических нарушений состояние оглушенности ступором, часто называемым прекоматозным состоянием, клинически проявляющимся глубоким сном или аналогичной ему по поведенческим реакциям ареактивностью. Конечной стадией нарастающего угнетения ЦНС является кома, характеризующаяся полным отсутствием сознания.
При исследовании крови определяются гипергликемия, гиперкетонемия, повышение уровня азота мочевины, креатинина и в ряде случаев – лактата. Уровень натрия в плазме обычно снижен. Несмотря на значительную потерю калия с осмотическим диурезом, рвотными массами и стулом, приводящую к выраженному дефициту данного электролита в организме, его концентрация в плазме может быть нормальной или даже слегка повышенной при анурии. При исследовании мочи определяются глюкозурия, кетонурия и протеинурия. Кислотно-основное состояние (КОС) отражает декомпенсированный метаболический ацидоз, причем в тяжелых случаях рН крови опускается ниже 7,0. На ЭКГ могут быть признаки гипоксии миокарда и нарушения проводимости.
При исследовании мочи определяются глюкозурия, кетонурия и протеинурия. Кислотно-основное состояние (КОС) отражает декомпенсированный метаболический ацидоз, причем в тяжелых случаях рН крови опускается ниже 7,0. На ЭКГ могут быть признаки гипоксии миокарда и нарушения проводимости.
В том случае, если известно о наличии у больного СД, диагностика кетоацидоза и кетоацидотической комы не представляет сложности. Диагноз подтверждается описанной выше клинической картиной, лабораторными показателями (прежде всего гипергликемией, наличием глюкозы и кетоновых тел в моче) и КОС, указывающими на наличие декомпенсированного метаболического ацидоза. В случае манифестации СД сразу в состоянии кетоацидоза или комы прежде всего следует ориентироваться на наличие выраженной дегидратации, признаков ацидоза (дыхание Куссмауля) и значительной потери массы тела за короткий отрезок времени. При этом исследование КОС исключает респираторный алкалоз как причину гипервентиляции и подтверждает наличие у больного метаболического ацидоза. Кроме того, запах ацетона в выдыхаемом воздухе должен навести врача на мысль о наличии у больного именно кетоацидоза. Лактат-ацидоз, уремия, алкогольный кетоацидоз, отравления кислотами, метанолом, этиленгликолем, паральдегидом, салицилатами (остальные причины метаболического ацидоза) не сопровождаются столь выраженной дегидратацией и значительной потерей массы тела, а также проявляются типичной для них клинической картиной. Наличие гипергликемии и кетонурии подтверждает диагноз СД и кетоацидоза.
Кроме того, запах ацетона в выдыхаемом воздухе должен навести врача на мысль о наличии у больного именно кетоацидоза. Лактат-ацидоз, уремия, алкогольный кетоацидоз, отравления кислотами, метанолом, этиленгликолем, паральдегидом, салицилатами (остальные причины метаболического ацидоза) не сопровождаются столь выраженной дегидратацией и значительной потерей массы тела, а также проявляются типичной для них клинической картиной. Наличие гипергликемии и кетонурии подтверждает диагноз СД и кетоацидоза.
Лечение
Лечение больных, находящихся в состоянии декомпенсации СД, а тем более в состоянии кетоацидоза или кетоацидотической комы, должно начинаться немедленно. Пациентов госпитализируют в специализированное отделение, а в состоянии комы – в отделение реанимации.
Основными целями терапии кетоацидоза являются борьба с дегидратацией и гиповолемическим шоком, восстановление физиологического КОС, нормализация электролитного баланса, ликвидация интоксикации и лечение сопутствующих заболеваний.
Непосредственно перед началом терапии больному промывают желудок раствором гидрокарбоната натрия. Для контроля функции почек и учета диуреза вводят мочевой катетер. С целью улучшения оксигенации тканей налаживают вдыхание кислорода. Учитывая гипотермию, больного необходмо тепло укрыть, а растворы следует вводить подогретыми.
Для контроля за эффективностью проводимой терапии до начала лечения контролируют гликемию, рН крови, рСО2, уровень К, Na, лактата и кетоновых тел в крови, глюкозурию и кетонурию, АД, ЭКГ, уровень гемоглобина, гематокрит, частоту дыхания (ЧД), пульс. В последующем необходимо ежечасно контролировать гликемию, рН крови, рСО2, АД, ЭКГ, ЧД, пульс. Оценивать прочие показатели можно каждые 2 – 3 ч.
Важное прогностическое значение (особенно в состоянии комы) имеет оценка реакции зрачков на свет. Слабая реакция или полное ее отсутствие свидетельствует о развившихся структурных изменениях в стволе мозга и низкой вероятности благоприятного исхода заболевания.
Регидратация очень важна в лечении диабетического кетоацидоза ввиду большой роли обезвоживания в цепочке метаболических расстройств при данном состоянии. Объем потерянной жидкости восполняется физиологическим (или гипотоническим при гиперосмолярности) и 5 – 10% растворами глюкозы. Прекращение инфузионной терапии возможно лишь при полном восстановлении сознания, отсутствии тошноты, рвоты и возможности приема жидкости больным per os. В течение первого часа внутривенно капельно вводят 1 л 0,9% раствора NaCl. При наличии гиперосмолярности физиологический раствор может быть заменен на гипотонический 0,45% раствор NaCl.
Эффективную осмолярность рассчитывают по следующей формуле:
Осмолярность = 2 [Na + K (ммоль/л)] + глюкоза крови (мОсм) (ммоль/л), нормальное значение = 297 ± 2 мОсм/л
В течение следующих двух часов от начала терапии ежечасно вводят по 500 мл 0,9% раствора NaCl. В последующие часы скорость введения жидкости обычно не должна превышать 300 мл/ч. После снижения уровня гликемии ниже 14 ммоль/л физиологический раствор заменяют на 5 – 10% раствор глюкозы и вводят со скоростью, указанной выше. Назначение глюкозы на данном этапе диктуется рядом причин, среди которых главная – поддержание необходимой осмолярности крови. Быстрое снижение уровня гликемии и концентрации прочих высокоосмолярных компонентов крови на фоне инфузионной терапии нередко становится причиной нежелательного быстрого снижения осмолярности плазмы.
После снижения уровня гликемии ниже 14 ммоль/л физиологический раствор заменяют на 5 – 10% раствор глюкозы и вводят со скоростью, указанной выше. Назначение глюкозы на данном этапе диктуется рядом причин, среди которых главная – поддержание необходимой осмолярности крови. Быстрое снижение уровня гликемии и концентрации прочих высокоосмолярных компонентов крови на фоне инфузионной терапии нередко становится причиной нежелательного быстрого снижения осмолярности плазмы.
Инсулинотерапию начинают сразу после постановки диагноза кетоацидоза. При лечении кетоацидоза, как и любого другого ургентного состояния при СД, используют инсулин только короткого действия (Актрапид МС, Актрапид НМ, Хумулин Р, Инсуман Рапид и др. ). До нормализации КОС и снижения уровня гликемии ниже 14,0 ммоль/л инсулин вводят только внутривенно капельно или внутримышечно в прямую мышцу живота. По достижении казанного уровня гликемии и нормализации КОС больного переводят на подкожное введение инсулина короткого действия.
Доза инсулина в первый час лечения составляет 10 ЕД внутривенно струйно или 20 ЕД внутримышечно. В случае сопутствующей тяжелой гнойной инфекции первую дозу инсулина можно увеличить вдвое.
В последующем ежечасно вводят в среднем по 6 ЕД инсулина короткого действия внутримышечно или вместе с физиологическим раствором NaСl внутривенно капельно. Для этого в отдельную емкость с 0,9% раствором NaCl добавляется 10 ЕД инсулина на каждые 100 мл физиологического раствора. Полученную смесь тщательно взбалтывают. С целью адсорбции инсулина на стенках системы через нее пропускают 50 мл смеси струйно. Применение для этой же цели использовавшихся ранее растворов альбумина в настоящее время считается необязательным. Ежечасно внутривенно капельно вводят 60 мл указанной смеси. В том случае, если в течение первых 2 – 3 ч от начала терапии уровень гликемии не снижается, дозу инсулина в последующий час рекомендуется удвоить.
При достижении уровня гликемии 12 – 14 ммоль/л дозу вводимого инсулина уменьшают в 2 раза – до 3 ЕД ежечасно (30 мл смеси инсулина и физиологического раствора). На данном этапе терапии возможен перевод больного на внутримышечные инъекции инсулина, однако следует иметь в виду, что используемые инсулиновые шприцы и различные индивидуальные системы для введения гормона снабжены иглами лишь для подкожного введения инсулина.
На данном этапе терапии возможен перевод больного на внутримышечные инъекции инсулина, однако следует иметь в виду, что используемые инсулиновые шприцы и различные индивидуальные системы для введения гормона снабжены иглами лишь для подкожного введения инсулина.
Не следует стремиться к снижению уровня гликемии ниже 10 ммоль/л, так как при этом возрастает риск не только гипогликемии, но и прежде всего – гипосмолярности. Тем не менее, если уровень гликемии снижается ниже 10 ммоль/л при сохраняющемся ацидозе, рекомендуется по-прежнему вводить инсулин ежечасно, а дозу снизить до 2 – 3 ЕД/ч. При нормализации КОС (легкая кетонурия при этом может сохраняться) следует перевести больного на подкожное введение инсулина по 6 ЕД каждые 2 ч, а затем – каждые 4 ч в той же дозе.
При отсутствии кетоацидоза на 2 – 3-и сутки лечения больной может быть переведен на 5 – 6 разовое введение инсулина короткого действия, а в дальнейшем – на обычную комбинированную инсулинотерапию.
Восстановление электролитного баланса, прежде всего дефицита калия, является важным компонентом комплексного лечения кетоацидоза. Обычно введение КCl начинают через 2 ч от начала инфузионной терапии. Однако если до начала лечения уже имеются ЭКГ- или лабораторные признаки, подтверждающие гипокалиемию при обязательном отсутствии анурии, введение калия можно начинать сразу, так как введение жидкости и инсулина способствует быстрому снижению уровня калия в крови за счет разведения его концентрации и нормализации транспорта калия в клетку.
Обычно введение КCl начинают через 2 ч от начала инфузионной терапии. Однако если до начала лечения уже имеются ЭКГ- или лабораторные признаки, подтверждающие гипокалиемию при обязательном отсутствии анурии, введение калия можно начинать сразу, так как введение жидкости и инсулина способствует быстрому снижению уровня калия в крови за счет разведения его концентрации и нормализации транспорта калия в клетку.
Доза раствора KCL, вводимого внутривенно капельно, зависит от концентрации калия в плазме. Так, при уровне калия ниже 3 ммоль/л необходимо вводить 3 г/ч (сухого вещества), при 3 – 4 ммоль/л – 2 г/ч, при 4 – 5 ммоль/л – 1,5 г/ч, при 5 – 6 ммоль/л – 0,5 г/час. По достижении уровня калия в плазме 6 ммоль/л введение раствора KCl следует прекратить.
Как правило, больные не нуждаются в дополнительной коррекции гипофосфатемии. Вопрос о необходимости введения фосфата калия возникает лишь в том случае, если уровень фосфора в плазме снижается ниже 1 мг%.
Восстановление КОС начинается буквально с первых минут лечения кетоацидоза, благодаря назначению жидкости и введению инсулина. Восстановление объема жидкости запускает физиологические буферные системы, в частности, восстанавливается способность почек реабсорбировать бикарбонаты. Назначение инсулина подавляет кетогенез и тем самым снижает концентрацию водородных ионов в крови. Однако в ряде случаев встает вопрос о необходимости назначения гидрокарбоната натрия с целью коррекции КОС. Выше было отмечено, что даже значительный периферический метаболический ацидоз далеко не всегда сопровождается столь же выраженным ацидозом ЦНС, благодаря наличию ряда защитно-приспособительных механизмов. По данным J. Ohman и соавт. J. Posner и F. Plum [2], у больных с диабетическим кетоацидозом до начала терапии рН спинномозговой жидкости обычно в пределах нормы. Попытки коррекции ацидоза плазмы с помощью внутривенного введения гидрокарбоната натрия могут привести к быстрому развитию ацидоза ЦНС и резкому ухудшению состояния сознания больного. С учетом описанных побочных явлений при введении соды разработаны очень жесткие критерии назначения гидрокарбоната натрия при диабетическом кетоацидозе.
Восстановление объема жидкости запускает физиологические буферные системы, в частности, восстанавливается способность почек реабсорбировать бикарбонаты. Назначение инсулина подавляет кетогенез и тем самым снижает концентрацию водородных ионов в крови. Однако в ряде случаев встает вопрос о необходимости назначения гидрокарбоната натрия с целью коррекции КОС. Выше было отмечено, что даже значительный периферический метаболический ацидоз далеко не всегда сопровождается столь же выраженным ацидозом ЦНС, благодаря наличию ряда защитно-приспособительных механизмов. По данным J. Ohman и соавт. J. Posner и F. Plum [2], у больных с диабетическим кетоацидозом до начала терапии рН спинномозговой жидкости обычно в пределах нормы. Попытки коррекции ацидоза плазмы с помощью внутривенного введения гидрокарбоната натрия могут привести к быстрому развитию ацидоза ЦНС и резкому ухудшению состояния сознания больного. С учетом описанных побочных явлений при введении соды разработаны очень жесткие критерии назначения гидрокарбоната натрия при диабетическом кетоацидозе. Рассматривать вопрос о целесообразности введения соды следует лишь при уровне рН крови ниже 7,0. Необходимо подчеркнуть, что при этом очень важно осуществлять постоянное мониторирование изменений КОС, а при достижении рН значения 7,0 введение гидрокарбоната следует прекратить. Используют 4% раствор гидрокарбоната натрия из расчета 2,5 мл на 1 кг фактической массы тела внутривенно капельно очень медленно. При введении гидрокарбоната натрия дополнительно внутривенно капельно вводят раствор KCl из расчета 1,5 – 2 г KCl сухого вещества [2].
Рассматривать вопрос о целесообразности введения соды следует лишь при уровне рН крови ниже 7,0. Необходимо подчеркнуть, что при этом очень важно осуществлять постоянное мониторирование изменений КОС, а при достижении рН значения 7,0 введение гидрокарбоната следует прекратить. Используют 4% раствор гидрокарбоната натрия из расчета 2,5 мл на 1 кг фактической массы тела внутривенно капельно очень медленно. При введении гидрокарбоната натрия дополнительно внутривенно капельно вводят раствор KCl из расчета 1,5 – 2 г KCl сухого вещества [2].
В целях лечения или профилактики воспалительных заболеваний назначаются антибиотики широкого спектра действия.
Для улучшения реологических свойств крови и предотвращения диссеминированного внутрисосудистого свертывания дважды в первые сутки лечения вводят 5000 ЕД гепарина внутривенно под контролем коагулограммы.
В целях нормализации окислительных процессов добавляют 150 – 200 мл кокарбоксилазы и 5 мл 5% раствора аскорбиновой кислоты.
При низком АД и других симптомах шока проводят терапию, направленную на повышение и поддержание АД и сердечной деятельности.
После выведения больного из состояния кетоацидоза назначают щадящую диету, богатую углеводами, белками, калием. Жиры из рациона питания исключают минимум на неделю.
Осложнения кетоацидоза
Среди осложнений, возникающих на фоне терапии кетоацидоза, наибольшую опасность представляет отек мозга, который в 70% случаев заканчивается летально (R. Couch и соавт., 1991; A. Glasgow, 1991). Наиболее частой причиной возникновения отека мозга является быстрое снижение осмолярности плазмы и уровня гликемии на фоне проводимой инфузионной терапии и введения инсулина. В случае применения гидрокарбоната натрия в целях коррекции ацидоза создаются дополнительные предпосылки для возникновения этого грозного осложнения. Дисбаланс между рН периферической крови и ликвора способствует повышению давления последнего и облегчает транспорт воды из межклеточного пространства в клетки мозга, осмолярность которых повышена. Обычно отек мозга развивается через 4 – 6 ч от начала терапии диабетического кетоацидоза. В том случае, когда сознание у больного сохранено, признаками начинающегося отека мозга являются ухудшение самочувствия, выраженная головная боль и головокружение, тошнота, рвота, расстройства зрения, а также напряжение глазных яблок, нестабильность гемодинамических показателей, нарастающая лихорадка. Как правило, перечисленные клинические симптомы появляются после “светлого” периода улучшения самочувствия на фоне очевидной положительной динамики лабораторных показателей.
Обычно отек мозга развивается через 4 – 6 ч от начала терапии диабетического кетоацидоза. В том случае, когда сознание у больного сохранено, признаками начинающегося отека мозга являются ухудшение самочувствия, выраженная головная боль и головокружение, тошнота, рвота, расстройства зрения, а также напряжение глазных яблок, нестабильность гемодинамических показателей, нарастающая лихорадка. Как правило, перечисленные клинические симптомы появляются после “светлого” периода улучшения самочувствия на фоне очевидной положительной динамики лабораторных показателей.
Значительно сложнее заподозрить начинающийся отек мозга у больных в состоянии кетоацидотической комы. Верным признаком данного осложнения на начальном этапе является отсутствие положительной динамики в состоянии сознания больного на фоне объективного улучшения показателей углеводного обмена. Описанные выше клинические признаки отека мозга сопровождаются снижением или отсутствием реакции зрачков на свет, офтальмоплегией и отеком зрительного нерва. Ультразвуковая энцефалография и компьютерная томография подтверждают диагноз.
Ультразвуковая энцефалография и компьютерная томография подтверждают диагноз.
Лечение отека мозга представляет значительно большие трудности, чем диагностика этого состояния. При подтверждении наличия у больного отека мозга назначают осмотические диуретики – внутривенное капельное введение раствора маннитола из расчета 1 – 2 г/кг. Вслед за этим внутривенно струйно вводят 80 – 120 мг лазикса и 10 мл гипертонического раствора натрия хлорида [4]. Вопрос о целесообразности назначения глюкокортикоидов (предпочтение отдается исключительно дексаметазону ввиду его минимальных минералокортикоидных свойств) до конца не решен. Считается, что наибольший эффект от назначения этих гормонов наблюдается при отеке мозга на фоне травмы или опухоли. Однако учитывая способность глюкокортикоидов снижать патологически повышенную проницаемость сосудов и гематоэнцефалического барьера, нормализовывать ионный транспорт через клеточную мембрану и тормозить активность лизосомальных ферментов клеток мозга, вопрос о целесообразности их назначения при отеке мозга при кетоацидозе следует решать индивидуально. К проводимым терапевтическим мероприятиям добавляются гипотермия мозга и активная гипервентиляция легких с целью снижения внутричерепного давления за счет возникающей при этом вазоконстрикции. В ряде случаев следует рассмотреть вопрос о проведении трепанации черепа.
К проводимым терапевтическим мероприятиям добавляются гипотермия мозга и активная гипервентиляция легких с целью снижения внутричерепного давления за счет возникающей при этом вазоконстрикции. В ряде случаев следует рассмотреть вопрос о проведении трепанации черепа.
Среди прочих осложнений кетоацидоза и его терапии следует выделить диссеминированное внутрисосудистое свертывание, отек легких, острую сердечно-сосудистую недостаточность, гипокалиемию, метаболический алкалоз, асфиксию вследствие аспирации желудочного содержимого.
Строгий контроль показателей гемодинамики, гемостаза, содержание электролитов, изменений осмолярности и неврологических симптомов позволяет заподозрить перечисленные выше осложнения на ранних стадиях и немедленно принять действенные меры, направленные на их ликвидацию.
Литература:
1. Krane E. Diabetic Ketoacidosis. Ped Clinics N Amer 1987;34:935–60.
2. Плам Ф., Познер Дж. Б. Диагностика ступора и комы. Пер.с англ.: Медицина, 1986. – 544 с. илл.
Б. Диагностика ступора и комы. Пер.с англ.: Медицина, 1986. – 544 с. илл.
3. Beaser R. Diabetic emergencies. Joslin Diabetes Center. Lecture Notes. October, 1992:12.
4. Diabetic ketoacidosis – A Scheme for management. In: Diabetes in the Young. ISGD. Official Bulletin 1990;23:13–5.
Синдром гиперкетонемии у детей и подростков: патогенез, причины, диагностика | #07/17
Часть 2. Начало статьи читайте в № 6, 2017 г.
Голодание
Голодание — это состояние организма, связанное с частичным или полным нарушением поступления пищи. В состоянии голодания резко снижаются источники энергии организма для важнейших структур организма. В условиях дефицита питательных веществ в организме образование энергии происходит за счет интенсификации глюкогенеза и синтеза кетоновых тел. Содержание глюкозы в крови уменьшается до нижних пределов нормы (3,5 ммоль/л) и на этом уровне поддерживается и в последующие периоды голодания. В печени при голодании глюкоза не в состоянии обеспечить должного количества оксалоацетата, поскольку ее просто нет в клетке. Поэтому при голодании жирные кислоты не «сгорают» в ЦТК, а превращаются в кетоновые тела.
Поэтому при голодании жирные кислоты не «сгорают» в ЦТК, а превращаются в кетоновые тела.
Снижение запасов гликогена в печени сопровождается усиленным поступлением в нее свободных жирных кислот из адипоцитов. Концентрация жирных кислот в крови увеличивается в 3–4 раза по сравнению с постабсорбтивным состоянием. Уровень кетоновых тел в крови через неделю голодания повышается в 10–15 раз. В то же время дефицит углеводов тормозит окисление кетоновых тел, замедляя ресинтез их в высшие жирные кислоты [13].
Энергетические потребности мышц и большинства других органов удовлетворяются за счет жирных кислот и кетоновых тел. При низком уровне инсулина глюкоза в мышечные клетки не проникает, потребителями глюкозы являются инсулинонезависимые клетки и прежде всего клетки мозга, но и в этой ткани биоэнергетика частично обеспечивается кетоновыми телами. При такой концентрации ацетоуксусная кислота активно декарбоксилируется с образованием ацетона, который выводится с выдыхаемым воздухом и через кожу. Уже на 3–4 день изо рта и от кожи голодающего исходит запах ацетона.
Уже на 3–4 день изо рта и от кожи голодающего исходит запах ацетона.
Организм включает альтернативные способы выработки энергии — это глюконеогенез и синтез кетокислот, которые потребляются центральной нервной системой. При голодании повышается выброс глюкагона, который активирует липолиз в адипоцитах и окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль клетки, где опять превращается в оксалоацетат и используется в глюконеогенезе.
Глюконеогенез продолжается за счет распада тканевых белков. Аминокислоты образуются в результате распада мышечных белков и включаются в глюконеогенез при длительном голодании. Пируват образуется в печени из лактата и аланина. Аланин и глутамин являются наиболее важными глюкогенными аминокислотами при голодании. Пируват и метаболиты ЦТК способны образовывать оксалоацетат и включаться в глюконеогенез.
При голодании подавляется использование ацетил-КоА в ЦТК, и он используется исключительно для синтеза оксиметилглутарил-КоА, что приводит к увеличению образования кетоновых тел. В этих условиях кетоновые тела являются альтернативным (глюкозе) энергетическим материалом для мозга и других тканей. 75% потребности мозга в энергии удовлетворяется за счет ацетил-КоА [4].
В этих условиях кетоновые тела являются альтернативным (глюкозе) энергетическим материалом для мозга и других тканей. 75% потребности мозга в энергии удовлетворяется за счет ацетил-КоА [4].
Если голодание продолжается дни, недели — включаются другие гомеостатические механизмы, которые обеспечивают сохранение белковой структуры организма, замедляя глюконеогенез и переключая мозг на утилизацию кетоновых молекул. Сигналом для использования кетонов служит повышение их концентрации в артериальной крови. При длительном голодании наблюдаются крайне низкие концентрации инсулина в крови. В этом случае интенсивный кетогенез представляет собой компенсаторно приспособительную реакцию.
Интенсивность обмена веществ в целом снижена: через неделю голодания потребление кислорода уменьшается примерно на 40%, происходят торможение окислительных процессов в митохондриях и угнетение окислительного фосфорилирования с образованием АТФ, т. е. развивается гипоэнергетическое состояние.
Накапливаясь в крови, кетоновые тела подавляют секрецию и активность глюкокортикоидов, тем самым препятствуя разрушению структурных белков организма и угнетая секрецию глюкагона [2]. Если в это время голодающему вводить аланин или другие гликогенные аминокислоты, уровень глюкозы в крови повышается, а концентрация кетоновых тел снижается.
Если в это время голодающему вводить аланин или другие гликогенные аминокислоты, уровень глюкозы в крови повышается, а концентрация кетоновых тел снижается.
При голодании кетоз опасности не представляет, так как не достигает степени кетоацидоза. Последний развивается при сопутствующих факторах — дегидратации, алкогольной интоксикации и других состояниях.
Алкогольная интоксикация
Гиперпродукция кетокислот и кетоацидоз после чрезмерного употребления спиртного частое наблюдаемое состояние. Катаболизм этилового спирта осуществляется главным образом в митохондриях печени. Здесь окисляется от 75% до 98% введенного в организм этанола. Окисление алкоголя — сложный биохимический процесс. Основную роль в метаболизме этанола играет никотинамидадениндинуклеотид (NAD). Этот фермент превращает этанол в токсический метаболит — ацетальдегид и восстановленный NADH, а последний соответствует синтезу ацетоацетата и β-оксибутирата.
Алкогольдегидрогеназа катализирует обратимую реакцию, направление которой зависит от концентрации ацетальдегида и соотношения NADH/NAD+ в клетке. Повышение концентрации ацетальдегида в клетке вызывает индукцию фермента альдегидоксидазы. В ходе реакции образуются уксусная кислота.
Повышение концентрации ацетальдегида в клетке вызывает индукцию фермента альдегидоксидазы. В ходе реакции образуются уксусная кислота.
Полученная в ходе реакции уксусная кислота активируется под действием фермента ацетил-КоА-синтетазы. Реакция протекает с использованием кофермента А и молекулы АТФ. Образовавшийся ацетил-КоА, в зависимости от соотношения АТФ/АДФ и концентрации оксалоацетата в митохондриях гепатоцитов, может «сгорать» в ЦТК или использоваться на синтез жирных кислот или кетоновых тел.
На начальных стадиях алкоголизма ацетил-КоА в ЦТК — основной источник энергии для клетки. Избыток ацетил-КоА в составе цитрата выходит из митохондрий, и в цитоплазме начинается синтез жирных кислот.
В период острой алкогольной интоксикации, несмотря на наличие большого количества ацетил-КоА, недостаток оксалоацетата снижает скорость образования цитрата. В этих условиях избыток ацетил-КоА идет на синтез кетоновых тел. Увеличение концентрации NADH по сравнению с NAD+ замедляет реакцию окисления лактата, увеличивается соотношение лактат/пируват. В крови возрастает концентрация лактата, это приводит к гиперлактацидемии и лактоацидозу. Повышение в крови содержания лактата, ацетоуксусной кислоты и β-гидроксибутирата служит причиной метаболического ацидоза при алкогольной интоксикации [14].
В крови возрастает концентрация лактата, это приводит к гиперлактацидемии и лактоацидозу. Повышение в крови содержания лактата, ацетоуксусной кислоты и β-гидроксибутирата служит причиной метаболического ацидоза при алкогольной интоксикации [14].
Способствует усиленному кетогенезу при алкогольной интоксикации гипогликемические состояния, связанные с рвотой и голоданием. Известно также, что у таких пациентов уровень инсулина в крови снижен, тогда как содержание кортизола, гормона роста, глюкагона и адреналина повышено. Этанол тормозит глюконеогенез. Дегидратация в этих случаях способствует кетогенезу.
Кетоз при нарушении гормональной регуляции
На уровень глюкозы в крови влияет широкий спектр гормонов, при этом только инсулин вызывает гипогликемический эффект. Контринсулярным действием с повышением уровня глюкозы крови обладают все гормоны: глюкагон, адреналин, глюкокортикоиды, адренокортикотропный (АКТГ), соматотропный (СТГ), тиреотропный (ТТГ), тиреоидные.
Эффекты инсулина и контринсулярных гормонов в норме регулируют стабильный уровень глюкозы в крови. При низкой концентрации инсулина усиливаются гипергликемические эффекты других гормонов, таких как глюкагон, адреналин, глюкокортикоиды и гормон роста. Это происходит даже в том случае, если концентрация этих гормонов в крови не увеличивается.
При низкой концентрации инсулина усиливаются гипергликемические эффекты других гормонов, таких как глюкагон, адреналин, глюкокортикоиды и гормон роста. Это происходит даже в том случае, если концентрация этих гормонов в крови не увеличивается.
Патогенез кетоза при избытке тироксина, глюкокортикоидов, соматотропина или/и других гормонов, в сущности, аналогичен уже рассмотренным механизмам гиперпродукции кетокислот вследствие избытка контринсулярных гормонов [6]. Известно, что в период усиленного роста, а также при гипертиреозе наступает значительное похудание.
Стресс
При стрессе активируется симпатическая нервная система и выброс контринсулярных гормонов, происходит истощение углеводных резервов организма, нарушается способность печени синтезировать и откладывать гликоген. Происходит избыточное поступление в печень неэтерифицированных жирных кислот. В результате повышенной продукции глюкокортикоидов идет распад белков и усиленное образование кетоновых тел из кетогенных аминокислот.
Гиперкортицизм
Ацетонемический синдром может быть первым клиническим проявлением гиперкортицизма, когда характерные признаки заболевания еще не сформировались.
Глюкокортикоиды способствуют усилению мобилизации нейтральных жиров из жировой ткани и тормозят липогенез. Но это действие в организме может перекрываться другими эффектами данных гормонов: способностью вызывать гипергликемию и стимулировать секрецию инсулина, накопление гликогена в печени, что приводит к торможению мобилизации жира и его отложению в жировой ткани; способностью в больших дозах задерживать жиромобилизующее и стимулирующее окисление жиров соматотропином.
Этим можно объяснить накопление жира в жировых депо при гиперкортицизме (болезни и синдроме Иценко–Кушинга). Кроме того, при этом состоянии увеличено образование дигидрокортизона, который стимулирует пентозный цикл и превращение углеводов в жиры. Кортикотропин, стимулируя секрецию глюкокортикоидов, может влиять на жировой обмен в том же направлении, но, помимо этого, обладает еще и экстраадреналовым жиромобилизующим действием [6].
Тиреотоксикоз
Избыток тиреоидных гормонов в крови может быть следствием заболеваний, проявляющихся гиперфункцией щитовидной железы. Тяжелым осложнением основного заболевания, сопровождающегося гиперфункцией щитовидной железы, является тиреотоксический криз, который представляет собой резкое обострение всех симптомов тиреотоксикоза. Чрезмерное поступление в кровь тироидных гормонов вызывает тяжелое токсическое поражение сердечно-сосудистой системы, печени, нервной системы и надпочечников. В клинической картине характерны резкое возбуждение (вплоть до психоза с бредом и галлюцинациями), которое затем сменяется адинамией, сонливостью, мышечной слабостью, апатией. Усиливаются диспепсические расстройства: жажда, тошнота, рвота, жидкий стул. Возможно увеличение печени. На этом фоне резко усиливаются процессы кетогенеза, что может спровоцировать симптомы ацетонемии.
Тироксин обладает жиромобилизующим эффектом. При гипертиреозе усилен обмен углеводов. Увеличена утилизация глюкозы тканями. Активируется фосфорилаза печени и мышц, следствием чего является усиление гликогенолиза и обеднение этих тканей гликогеном. Увеличивается активность гексокиназы и всасывание глюкозы в кишечнике, что может сопровождаться алиментарной гипергликемией. Активируется инсулиназа печени, что вместе с гипергликемией вызывает напряженное функционирование инсулярного аппарата и в случае его функциональной неполноценности может привести к развитию сахарного диабета. Усиление пентозного пути обмена углеводов способствует образованию НАДФ-Н2. В надпочечниках это вызывает повышение стероидогенеза и большее образование кортикостероидов [4].
Увеличена утилизация глюкозы тканями. Активируется фосфорилаза печени и мышц, следствием чего является усиление гликогенолиза и обеднение этих тканей гликогеном. Увеличивается активность гексокиназы и всасывание глюкозы в кишечнике, что может сопровождаться алиментарной гипергликемией. Активируется инсулиназа печени, что вместе с гипергликемией вызывает напряженное функционирование инсулярного аппарата и в случае его функциональной неполноценности может привести к развитию сахарного диабета. Усиление пентозного пути обмена углеводов способствует образованию НАДФ-Н2. В надпочечниках это вызывает повышение стероидогенеза и большее образование кортикостероидов [4].
Дефицит гормонов
Гипогликемия всегда встречается при пангипопитуитаризме — заболевании, характеризующемся снижением и выпадением функции передней доли гипофиза (секреции адренокортикотропина, пролактина, соматотропина, фоллитропина, лютропина, тиреотропина). В результате резко снижается функция периферических эндокринных желез. Однако гипогликемия встречается и при первичном поражении эндокринных органов (врожденная дисфункция коры надпочечников, болезнь Аддисона, гипотиреоз, гипофункция мозгового слоя надпочечников, дефиците глюкагона). При дефиците контринсулярных гормонов снижается скорость глюконеогенеза в печени (влияние на синтез ключевых ферментов), повышается утилизация глюкозы на периферии, снижается образование аминокислот в мышцах — субстрата для глюконеогенеза.
Однако гипогликемия встречается и при первичном поражении эндокринных органов (врожденная дисфункция коры надпочечников, болезнь Аддисона, гипотиреоз, гипофункция мозгового слоя надпочечников, дефиците глюкагона). При дефиците контринсулярных гормонов снижается скорость глюконеогенеза в печени (влияние на синтез ключевых ферментов), повышается утилизация глюкозы на периферии, снижается образование аминокислот в мышцах — субстрата для глюконеогенеза.
Дефицит глюкокортикоидов
Первичная надпочечниковая недостаточность является следствием уменьшения секреции гормонов коры надпочечников. Под этим термином подразумевают различные по этиологии и патогенезу варианты гипокортицизма. Симптомы надпочечниковой недостаточности развиваются только после разрушения 90% объема ткани надпочечников.
Причины гипогликемии при надпочечниковой недостаточности схожи с причинами гипогликемии при гипопитуитаризме. Отличием является уровень возникновения блока — при гипопитуитаризме снижается секреция кортизола из-за дефицита АКТГ, а при надпочечниковой недостаточности из-за разрушения ткани самих надпочечников.
Гипогликемические состояния у больных с хронической надпочечниковой недостаточностью могут возникать как натощак, так и через 2–3 часа после приема пищи, богатой углеводами. Приступы сопровождаются слабостью, чувством голода, потливостью. Гипогликемия развивается в результате снижения секреции кортизола, уменьшения глюконеогенеза, запасов гликогена в печени.
Дефицит катехоламинов
Данное состояние может возникать при надпочечниковой недостаточности с поражением мозгового слоя надпочечников. Катехоламины, попадая в кровь, регулируют высвобождение и метаболизм инсулина, снижая его, а также увеличивают высвобождение глюкагона. При снижении секреции катехоламинов наблюдаются гипогликемические состояния, вызванные избыточной продукцией инсулина и пониженной активностью гликогенолиза.
Дефицит глюкагона
Глюкагон — гормон, являющийся физиологическим антагонистом инсулина. Он участвует в регуляции углеводного обмена, влияет на жировой обмен, активируя ферменты, расщепляющие жиры. Основное количество глюкагона синтезируется альфа-клетками островков поджелудочной железы. Однако установлено, что специальные клетки слизистой оболочки двенадцатиперстной кишки и слизистой оболочки желудка также синтезируют глюкагон. При поступлении в кровоток глюкагон вызывает повышение в крови концентрации глюкозы, вплоть до развития гипергликемии. В норме глюкагон предотвращает чрезмерное снижение концентрации глюкозы. Благодаря существованию глюкагона, препятствующего гипогликемическому действию инсулина, достигается тонкая регуляция обмена глюкозы в организме.
Основное количество глюкагона синтезируется альфа-клетками островков поджелудочной железы. Однако установлено, что специальные клетки слизистой оболочки двенадцатиперстной кишки и слизистой оболочки желудка также синтезируют глюкагон. При поступлении в кровоток глюкагон вызывает повышение в крови концентрации глюкозы, вплоть до развития гипергликемии. В норме глюкагон предотвращает чрезмерное снижение концентрации глюкозы. Благодаря существованию глюкагона, препятствующего гипогликемическому действию инсулина, достигается тонкая регуляция обмена глюкозы в организме.
При дефиците вышеперечисленных гормонов содержание инсулина снижено, а экскреция кетоновых тел с мочой повышена [4].
Роль печени в нарушении энергетического обмена
Печень участвует в поддержании нормального уровня глюкозы в сыворотке крови путем гликогеногенеза, гликогенолиза и глюконеогенеза. В основе нарушений обмена углеводов при болезнях печени лежат повреждения митохондрий, которые ведут к снижению окислительного фосфорилирования. Вторично страдают функции печени. При тяжелом остром гепатите, как правило, отмечается гипогликемия, а при циррозах печени это наступает в конечной стадии — при печеночной недостаточности [15]. Гипогликемия объясняется снижением способности печени (из-за обширного поражения ее паренхимы) синтезировать гликоген и уменьшением выработки инсулиназы (фермента, разрушающего инсулин).
Вторично страдают функции печени. При тяжелом остром гепатите, как правило, отмечается гипогликемия, а при циррозах печени это наступает в конечной стадии — при печеночной недостаточности [15]. Гипогликемия объясняется снижением способности печени (из-за обширного поражения ее паренхимы) синтезировать гликоген и уменьшением выработки инсулиназы (фермента, разрушающего инсулин).
Дефицит углеводов приводит также к усилению анаэробного гликолиза, вследствие чего в клетках накапливаются кислые метаболиты, вызывающие снижение рН. При циррозе печени может повышаться и уровень лактата в сыворотке крови в связи со сниженной способностью печени утилизировать его для глюконеогенеза.
При заболеваниях печени увеличивается роль жиров в качестве источника энергии. В печени происходят синтез жирных кислот и их расщепление до ацетил-КоА, а также образование кетоновых тел, насыщение ненасыщенных жирных кислот и их включение в ресинтез нейтральных жиров и фосфолипидов. Катаболизм жирных кислот осуществляется путем β-окисления, основной реакцией которого является активирование жирной кислоты с участием кофермента ацетил-КоА и АТФ. Освобождающийся ацетил-КоА подвергается полному окислению в митохондриях, в результате чего клетки обеспечиваются энергией.
Освобождающийся ацетил-КоА подвергается полному окислению в митохондриях, в результате чего клетки обеспечиваются энергией.
При ряде заболеваний печени снижается и синтез липопротеидов, что ведет к накоплению триацилглицеридов с последующей инфильтрацией и жировой дистрофией печени. Причинами возникновения этого состояния, в частности, является недостаток в пище липотропных веществ (холина — составной части лецитина, метионина). Увеличивается образование кетоновых тел [4].
Итак, клиническая картина вторичного ацетонемического синдрома включает в себя непосредственно явления кетоза, признаки основного заболевания, на фоне которого развился кетоз, а также проявления того состояния, которое запустило патологический процесс (стресс, чрезмерная физическая нагрузка, инфекция и т. д.).
Ацетонемическая циклическая рвота
В практике приходится сталкиваться с идиопатической ацетонемической рвотой, которая протекает с кетоацидозом (ацетонемическая рвота, недиабетический кетоацидоз). В англоязычной литературе она входит в синдром идиопатической циклической рвоты [16, 17].
В англоязычной литературе она входит в синдром идиопатической циклической рвоты [16, 17].
Патогенез ацетонемической рвоты полностью не выяснен. Предполагается, что у детей после перенесенных инфекционных заболеваний, травм черепа, органических заболеваний центральной нервной системы в течение длительного времени в гипоталамо-диэнцефальной области остается доминантный очаг застойного возбуждения, индуцирующий нарушения жирового обмена (усиление кетогенеза, нарушение нормального использования кетоновых тел в связи с истощением углеводных запасов в организме). В патогенезе ацетонемической рвоты могут иметь значение аномалии конституции, относительная несостоятельность энзимных систем печени, нарушения эндокринной регуляции метаболизма.
Перспективными являются представления о синдроме циклической рвоты как о митохондриальной патологии [18, 19]. Поскольку митохондрии являются, образно выражаясь, энергетическими станциями клетки, при данном заболевании нарушается энергетический обмен. В условиях стресса и гипоксии энергетический обмен нарушается с преобладанием более быстрого анаэробного гликолиза, но при этом образуется только 2 молекулы АТФ, тогда как при аэробном — 38 [5]. Возникает дефицит энергии.
В условиях стресса и гипоксии энергетический обмен нарушается с преобладанием более быстрого анаэробного гликолиза, но при этом образуется только 2 молекулы АТФ, тогда как при аэробном — 38 [5]. Возникает дефицит энергии.
Такие нарушения тесно связаны с нарушениями пуринового обмена, поскольку энергия в организме хранится в виде нуклеотидов, среди которых аденин и гуанин являются пуриновыми, и они метаболизируются до мочевой кислоты, а тимин, цитозин и урацил являются пиримидиновыми и метаболизируются с образованием кетоновых тел, аммиака и β-изомасляной кислоты. Данные представления патогенетически сближают синдром циклической рвоты и синдром ацетонемической рвоты, а также объясняют необходимость и возможные пути метаболической коррекции.
Другие считают, что причиной резкого повышения кетоновых тел может быть недостаточное потребление детьми углеводов при избытке жиров и кетогенных аминокислот.
Кризы могут возникать внезапно с промежутками в несколько недель или месяцев. Провоцирующими факторами могут быть: нарушение диеты (жареные и печеные продукты), лихорадка, отказ от еды, физические и психические перегрузки.
Провоцирующими факторами могут быть: нарушение диеты (жареные и печеные продукты), лихорадка, отказ от еды, физические и психические перегрузки.
Предвестниками синдрома циклической рвоты является анорексия, вялость или повышение возбудимости, тошнота, головные боли, абдоминальные боли, запах ацетона изо рта.
Затем появляется многократная или неукротимая рвота, которая может продолжаться от одного до пяти дней. Схваткообразные боли в животе усиливаются. Во время криза больной становится сонливым. В результате рвоты могут развиваться гемодинамические нарушения: тахикардия, мягкий пульс, приглушенность сердечных тонов, гипотония.
Печень умеренно увеличена. В некоторых случаях повышается температура. В выдыхаемом воздухе и рвотных массах ощущается запах прелых яблок. В моче высокая концентрация кетоновых тел. Приступы могут ликвидироваться спонтанно, без лечения.
Избыток кетоновых тел оказывает наркотическое действие на центральную нервную систему, что клинически проявляется вялостью, заторможенностью.
В биохимическом анализе крови обнаруживают нарушение липидного обмена (гиперхолестеринемию), тенденцию к гипогликемии, гиперкетонемию. В общем анализе крови: умеренный лейкоцитоз, нейтрофилез, ускоренная СОЭ.
В моче и выдыхаемом воздухе обнаруживается ацетон, в крови — повышенная концентрация кетоновых тел. На электроэнцефалограмме выявляются различные отклонения, не исчезающие полностью после прекращения приступа.
Этот синдром чаще встречается в дошкольном возрасте и сопровождается приступами многократной рвоты и кетонемии. У таких больных нередко выявляют повышенную возбудимость, мочекислую нефропатию, сахарный диабет, ожирение.
Кетоз при длительной рвоте, недоедании или голодании представляет классический компенсаторный процесс, призванный восполнить энергетический дефицит, точнее, недостаток углеводов, за счет альтернативных энергосубстратов кетокислот.
Диагноз синдрома ацетонемической рвоты можно подтвердить только после исключения других заболеваний, сопровождающихся рвотой: аппендицита и перитонита, энцефалитов, менингитов, начала отека головного мозга, отравления, токсикоза и инфекционных заболеваний и др. Но в первую очередь диабетического кетоацидоза.
Но в первую очередь диабетического кетоацидоза.
Ацетонемические кризы у большинства детей прекращаются после 10–12 лет, но сохраняется большая вероятность развития таких патологических состояний, как подагрические кризы, вегетососудистые дистонии по гипертоническому типу, артериальная гипертензия.
Транзиторный кетоз у детей и подростков может выявляться при лихорадке, стрессах, инфекционных заболеваниях, голодании (во время болезни), употреблении богатой жирами пищи, напряженной физической активности. В этих случаях содержания кетоновых тел в моче не более 2+.
Лечение
Лечение и профилактика гиперкетонемии зависят от причины ее возникновения, но во всех случаях направлены на улучшение функции печени и нормализацию энергетического обмена. Это достигается ограничением содержания жира в пищевом рационе, назначением липотропных средств (метионина и др.), витаминов группы В, при необходимости — инсулина, кокарбоксилазы.
В период приступа синдрома циклической ацетонемической рвоты выраженная дегидратация, гиповолемия, метаболический ацидоз и электролитные нарушения — это основные факторы, которые определяют тяжесть состояния. Необходимо в первую очередь ликвидировать ацидоз: назначить промывание желудка и кишечника 1–2% раствором бикарбоната натрия. Антикетогенными свойствами обладает 5–10% раствор глюкозы, с добавлением необходимого количества инсулина, а также раствор Рингера [20].
Необходимо в первую очередь ликвидировать ацидоз: назначить промывание желудка и кишечника 1–2% раствором бикарбоната натрия. Антикетогенными свойствами обладает 5–10% раствор глюкозы, с добавлением необходимого количества инсулина, а также раствор Рингера [20].
Если питье не провоцирует рвоту, рекомендуется подслащенный чай, Регидрон, Оралит — частыми и небольшими объемами. После улучшения состояния и появления возможности приема жидкости назначается кормление ребенка. Диета должна содержать легкоусвояемые углеводы и ограниченное количество жиров (манная, овсяная, гречневая каши; картофельное пюре, печеные яблоки, сухари, сухое печенье).
Итак, выяснение механизмов развития кетонемического синдрома, выделение наиболее вероятных причин формирования кетоза дают возможность установить генез заболевания, а тем самым нормализовать состояние больного и предупреждать рецидивы кетонемии.
Литература
- Березов Т. Т., Коровкин Б. Ф. Биологическая химия.
 Учебник. 3-е изд. М.: Медицина, 2004. 704 с.
Учебник. 3-е изд. М.: Медицина, 2004. 704 с. - Stryer, Lubert. Biochemistry (Fourth ed.). New York: W. H. Freeman and Company, 1995. P. 510–515, 581–613, 775–778.
- Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. Пер. с англ. М.: Мир, 1993. Т. I. 381 с.
- Зайчик А. Ш., Чурилов Л. П. Основы патохимии. СПб: Элби-СПб, 2000. 687 с.
- Эндокринология и метаболизм. В 2 т. / Под ред. Фелинга Ф. и соавт. Пер. с англ. Кандрора В. И., Старковой Н. Т. М.: Медицина,1985. Т. 2. 416 с.
- Эндокринология: национальное руководство / Под ред. Дедова И. И., Мельниченко Г. А. М.: ГЭОТАР-Медиа, 2008.
- Лечение диабетической комы у детей. Методические рекомендации. М., 2006. 14 с.
- Brown L. M., Corrado M. M., van der Ende R. M. et al. Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children // J Inherit Metab Dis. 2015, May; 38 (3): 489–493.

- Чибирас П. П. Гипогликемическая кетонемия как причина нейротоксикоза у детей // Вопросы охраны материнства и детства. 1982. № 2. С. 30–33.
- Генес С. Г. Гипогликемии. Гипогликемический симптомокомплекс. М.: Медицина, 1970. 236 с.
- Кроненберг Г. М. и соавт. Ожирение и нарушение липидного обмена. Пер. с англ. под ред. И. И. Дедова, Г. А. Мельниченко. М.: ООО «Рид Элсивер», 2010. 264 с.
- Лукьянчиков В. С. Кетоз и кетоацидоз. Патохимический и клинический аспект // РМЖ, 2004, т. 12, № 23, с. 1301.
- Масловская А. А. Механизм развития кетоза при сахарном диабете и голодании // Журнал Гродненского государственного медицинского университета. 2012, № 3 (39), 8–10.
- Неотложная медицинская помощь. Пер. с англ. / Под ред. Дж. Э. Тинтиналли, Р. Л. Кроума, Э. Руиза. М.: Медицина, 2001. 1000 c.
- Рябчук Ф. Н., Пирогова З. И. Коэнзимное ацетилирование и уровень свободных жирных кислот крови у детей с ацетонемией и билиарной недостаточностью // Лечащий Врач.
 2012, № 8, с. 42–46.
2012, № 8, с. 42–46. - Li BUK: Cyclic vomiting: new understanding of an old disorder // Contemporary Ped. 1996, 13 (7): 48–62.
- Krakowczyk H., Machura E., Rusek-Zychma M., Chrobak E., Ziora K. Assessment of the natural history and clinical presentation of acetonemic vomiting. 2014, 71 (6): 323–327.
- Boles R. et al. Cyclic vomiting syndrome and mitochondrial DNA mutations // Lancet. 1997, 350: 1299–1300.
- Salpietro C. D., Briuglia S., Merlino M. V. et al. A mitochondrial DNA mutation (A3243 G mtDNA) in a family with cyclic vomiting // Am. J. Pediatr. 2003. 162. 727–728.
- Марушко Ю. В., Шев Г. Г., Полковниченко Л. Н., Мошкина Т. В. Терапевтические подходы при ацетонемическом синдроме у детей // Здоровье ребенка. 2012, № 1, с. 61–65.
В. В. Смирнов1, доктор медицинских наук, профессор
А. В. Симаков
ФГБОУ ВО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва
Н. И. Пирогова МЗ РФ, Москва
1 Контактная информация: [email protected]
Детский диабетический кетоацидоз (ДКА): основы практики, фон, патофизиология
McGregor S, Metzger DL, Amed S, Goldman RD. Управление инфузионной системой у детей с диабетическим кетоацидозом. Can Fam Врач . 2020 Ноябрь 66 (11): 817-9. [Ссылка QxMD MEDLINE]. [Полный текст].
Эдж Дж. А., Рой Ю., Бергоми А. и др. Уровень сознания у детей с диабетическим кетоацидозом связан с тяжестью ацидоза, а не с концентрацией глюкозы в крови. Педиатрический диабет . 2006 7 февраля (1): 11-5. [Ссылка QxMD MEDLINE].
Harris GD, Fiordalisi I. Физиологическое лечение диабетической кетоацидемии. 5-летний проспективный педиатрический опыт, 231 эпизод. Arch Pediatr Adolesc Med .
 1994 г., октябрь 148 (10): 1046-52. [Ссылка QxMD MEDLINE].
1994 г., октябрь 148 (10): 1046-52. [Ссылка QxMD MEDLINE].Wolfsdorf J, Craig ME, Daneman D, et al. Диабетический кетоацидоз. Педиатрический диабет . 2007 8 февраля (1): 28-43. [Ссылка QxMD MEDLINE].
Маршалл С.М., Уокер М., Альберти КГММ. Диабетический кетоацидоз и гипергликемическая некетотическая кома. Альберти, Зиммет, Дефронцо, ред. Международный учебник по сахарному диабету . 1997. 1215-30.
Fagan MJ, Avner J, Khine H. Первоначальная инфузионная терапия у пациентов с диабетическим кетоацидозом: насколько они сухие? Клин Педиатр (Фила) . 2008 ноябрь 47(9):851-5. [Ссылка QxMD MEDLINE].
Durr JA, Hoffman WH, Sklar AH, et al. Корреляты отека головного мозга при неконтролируемом ИЗСД. Диабет . 1992 май.
 41(5):627-32. [Ссылка QxMD MEDLINE].
41(5):627-32. [Ссылка QxMD MEDLINE].Хейл П.М., Резвани И., Браунштейн А.В. и др. Факторы, предсказывающие отек головного мозга у детей раннего возраста с диабетическим кетоацидозом и впервые выявленным диабетом I типа. Акта Педиатр . 1997 июнь 86(6):626-31. [Ссылка QxMD MEDLINE].
Мел Дж.М., Вертер Г.А. Частота и исход диабетического отека мозга в детском возрасте: есть ли предикторы? J Педиатр Детское здоровье . 1995 31 февраля (1): 17-20. [Ссылка QxMD MEDLINE].
Silver SM, Clark EC, Schroeder BM, Sterns RH. Патогенез отека головного мозга после лечения диабетического кетоацидоза [опубликованные опечатки появляются в Kidney Int 1997 May; 51(5):1662]. Почки Внутренний . 1997 г., апрель 51(4):1237-44. [Ссылка QxMD MEDLINE].
 webmd.com»> Okuda Y, Adrogue HJ, Field JB и др. Контрпродуктивные эффекты бикарбоната натрия при диабетическом кетоацидозе. J Клин Эндокринол Метаб . 1996 янв. 81(1):314-20. [Ссылка QxMD MEDLINE].
webmd.com»> Okuda Y, Adrogue HJ, Field JB и др. Контрпродуктивные эффекты бикарбоната натрия при диабетическом кетоацидозе. J Клин Эндокринол Метаб . 1996 янв. 81(1):314-20. [Ссылка QxMD MEDLINE].Азова С., Рапапорт Р., Вольфсдорф Дж. Черепно-мозговая травма у детей с диабетическим кетоацидозом: обзор литературы и предполагаемый патофизиологический путь развития отека мозга. Педиатрический диабет . 2020 16 ноября. [Ссылка QxMD MEDLINE].
Глейзер Н. Церебральная травма и отек головного мозга у детей с диабетическим кетоацидозом: могут ли иметь место церебральная ишемия и реперфузионное повреждение?. Педиатрический диабет . 2009 г. 10 (8): 534-41 декабря. [Ссылка QxMD MEDLINE].
Musey VC, Lee JK, Crawford R. Диабет у городских афроамериканцев. I. Прекращение инсулинотерапии является основной причиной диабетического кетоацидоза.
 Лечение диабета . 1995 г. 18 апреля (4): 483-9. [Ссылка QxMD MEDLINE].
Лечение диабета . 1995 г. 18 апреля (4): 483-9. [Ссылка QxMD MEDLINE].Томпсон С.Дж., Каммингс Ф., Чалмерс Дж., Ньютон Р.В. Аномальное поведение при лечении инсулином: основная причина кетоацидоза у молодых людей. Диабет Мед . 1995 май. 12(5):429-32. [Ссылка QxMD MEDLINE].
Моррис А.Д., Бойл Д.И., МакМахон А.Д. и др. Приверженность к лечению инсулином, гликемический контроль и кетоацидоз при инсулинозависимом сахарном диабете. Сотрудничество DARTS/MEMO. Аудит и исследования диабета в Tayside, Шотландия. Отдел мониторинга лекарственных средств. Ланцет . 1997, 22 ноября. 350(9090):1505-10. [Ссылка QxMD MEDLINE].
Смальдоне А., Хониг Дж., Стоун П.В. и др. Характеристики калифорнийских детей с однократной и множественной госпитализацией по поводу диабетического кетоацидоза (1998-2000).
 Лечение диабета . 2005 г. 28 августа (8): 2082-4. [Ссылка QxMD MEDLINE]. [Полный текст].
Лечение диабета . 2005 г. 28 августа (8): 2082-4. [Ссылка QxMD MEDLINE]. [Полный текст].Holstein A, Abel C, Zumwalde I. Рецидивирующий тяжелый диабетический кетоацидоз вследствие интоксикации синтетическими наркотиками («Экстази» и «Спид»). Intensivmedizin и Notfallmedizin . 1997. 34(1):46-50.
Реверс А., Клингенсмит Г., Дэвис С. и др. Наличие диабетического кетоацидоза при диагностике сахарного диабета у молодежи: поиск диабета в молодежном исследовании. Педиатрия . 2008 май. 121(5):e1258-66. [Ссылка QxMD MEDLINE].
Smith CP, Firth D, Bennett S, et al. Кетоацидоз у детей с недавно диагностированным и установленным диабетом. Акта Педиатр . 1998 май. 87(5):537-41. [Ссылка QxMD MEDLINE].
Реверс А., Чейз Х.
 П., Маккензи Т. и др. Предикторы острых осложнений у детей с сахарным диабетом 1 типа. ЯМА . 2002 г., 15 мая. 287(19):2511-8. [Ссылка QxMD MEDLINE].
П., Маккензи Т. и др. Предикторы острых осложнений у детей с сахарным диабетом 1 типа. ЯМА . 2002 г., 15 мая. 287(19):2511-8. [Ссылка QxMD MEDLINE].Леви-Маршал С., Паттерсон С.С., Грин А. Географические различия при диагностике диабета I типа у детей: исследование EURODIAB. Европейский и Дибетес. Диабетология . 2001 Октябрь 44 Дополнение 3: B75-80. [Ссылка QxMD MEDLINE].
Edge JA, Dunger DB. Варианты лечения диабетического кетоацидоза у детей. Диабет Мед . 1994 г., 11 декабря (10): 984-6. [Ссылка QxMD MEDLINE].
Ной А., Хофер С.Е., Каргес Б., Оверинк Р., Розенбауэр Дж., Холл Р.В. Кетоацидоз в дебюте диабета все еще часто встречается у детей и подростков: многоцентровый анализ 14 664 пациентов из 106 учреждений. Лечение диабета . 2009 г. 32 сентября (9): 1647-8.
 [Ссылка QxMD MEDLINE]. [Полный текст].
[Ссылка QxMD MEDLINE]. [Полный текст].Usher-Smith JA, Thompson MJ, Sharp SJ, Walter FM. Факторы, связанные с наличием диабетического кетоацидоза при диагностике диабета у детей и молодых людей: систематический обзор. БМЖ . 2011 г., 7 июля. 343: d4092. [Ссылка QxMD MEDLINE].
Фрич М., Розенбауэр Дж., Шобер Э., Ной А., Плачек К., Холл Р.В. Предикторы диабетического кетоацидоза у детей и подростков с сахарным диабетом 1 типа. Опыт работы с большой многоцентровой базой данных. Педиатрический диабет . 2011 12 июня (4 часть 1): 307-12. [Ссылка QxMD MEDLINE].
Деламатер А.М., Шоу К.Х., Эпплгейт Э.Б. и др. Риск проблем с метаболическим контролем у молодежи из числа меньшинств с диабетом. Лечение диабета . 1999 май. 22(5):700-5. [Ссылка QxMD MEDLINE]. [Полный текст].
 webmd.com»> Кон Б.А., Сирилло П.М., Вингард Д.Л. и др. Гендерные различия в госпитализации по поводу ИЗСД среди подростков в Калифорнии, 1991 г. Значение для профилактики. Лечение диабета . 1997 20 ноября (11): 1677-82. [Ссылка QxMD MEDLINE].
webmd.com»> Кон Б.А., Сирилло П.М., Вингард Д.Л. и др. Гендерные различия в госпитализации по поводу ИЗСД среди подростков в Калифорнии, 1991 г. Значение для профилактики. Лечение диабета . 1997 20 ноября (11): 1677-82. [Ссылка QxMD MEDLINE].Куинн М., Флейшман А., Рознер Б. и др. Особенности диагностики сахарного диабета 1 типа у детей до 6 лет. J Педиатр . 2006 март 148(3):366-71. [Ссылка QxMD MEDLINE].
Гетти С., Ли Дж. К., Симс К. Э., Демастер Д. М., Глейзер Н. С. Диабетический кетоацидоз и нарушение памяти у детей с сахарным диабетом 1 типа. J Педиатр . 2010 янв. 156(1):109-14. [Ссылка QxMD MEDLINE].
Edge JA, Ford-Adams ME, Dunger DB и др. Причины смерти детей с инсулинозависимым диабетом 1990-96 гг. Арч Ди Чайлд . 1999 г., октябрь 81 (4): 318–323. [Ссылка QxMD MEDLINE].
 [Полный текст].
[Полный текст].Ной А., Виллаш А., Эххальт С. и др. Кетоацидоз в дебюте сахарного диабета 1 типа у детей — частота и клиническая картина. Педиатрический диабет . 2003 июнь 4 (2): 77-81. [Ссылка QxMD MEDLINE].
Warner DP, McKinney PA, Law GR, Bodansky HJ. Смертность и диабет из популяционного регистра в Йоркшире, 1978-93 гг. Арч Ди Чайлд . 1998 май. 78(5):435-8. [Ссылка QxMD MEDLINE]. [Полный текст].
Hoffman WH, Locksmith JP, Burton EM и др. Интерстициальный отек легких у детей и подростков с диабетическим кетоацидозом. J Осложнения диабета . 1998 ноябрь-декабрь. 12(6):314-20. [Ссылка QxMD MEDLINE].
Holsclaw DS Jr, Torcato B. Острый отек легких при ювенильном диабетическом кетоацидозе. Детский пульмонол .
 1997 г., 24 декабря (6): 438-43. [Ссылка QxMD MEDLINE].
1997 г., 24 декабря (6): 438-43. [Ссылка QxMD MEDLINE].Ян Э.М., Ли Х.Г., О К.И., Ким С.Дж. Острое повреждение почек при диабетическом кетоацидозе у детей. Индийский J Педиатр . 2020 19 ноября. [Ссылка QxMD MEDLINE].
Майерс С.Р., Глейзер Н.С., Трейнор Дж.Л. и др., Сеть прикладных исследований педиатрической неотложной помощи (PECARN) Исследовательская группа DKA FLUID. Частота и факторы риска острого повреждения почек при диабетическом кетоацидозе у детей и связь с нейрокогнитивными исходами. Открытие сети JAMA . 1 декабря 2020 г. 3 (12): e2025481. [Ссылка QxMD MEDLINE]. [Полный текст].
Wei Y, Wu C, Su F, Zhang H, Zhang J, Zheng R. Клинические характеристики и исходы у пациентов с диабетическим кетоацидозом различной степени тяжести. Медицина (Балтимор) . 2020 6 нояб.
 99 (45): e22838. [Ссылка QxMD MEDLINE]. [Полный текст].
99 (45): e22838. [Ссылка QxMD MEDLINE]. [Полный текст].Мьюир А.Б., Квислинг Р.Г., Ян М.С., Розенблюм А.Л. Отек головного мозга при диабетическом кетоацидозе у детей: естественное течение, рентгенологические данные и раннее выявление. Лечение диабета . 2004 г. 27 июля (7): 1541-6. [Ссылка QxMD MEDLINE].
Sottosanti M, Morrison GC, Singh RN, Sharma AP, Fraser DD, Alawi K, et al. Дегидратация у детей с диабетическим кетоацидозом: проспективное исследование. Arch Dis Child . 2012 фев. 97(2):96-100. [Ссылка QxMD MEDLINE].
Бранденбург, Массачусетс, Dire DJ. Сравнение значений газов артериальной и венозной крови при первичной оценке пациентов с диабетическим кетоацидозом в отделении неотложной помощи. Энн Эмерг Мед . 1998 г. 31 апреля (4): 459-65. [Ссылка QxMD MEDLINE].
 webmd.com»> Wiggam MI, O’Kane MJ, Harper R, et al. Лечение диабетического кетоацидоза с нормализацией концентрации 3-гидроксибутирата в крови в качестве конечной точки неотложной помощи. Рандомизированное контролируемое исследование. Лечение диабета . 1997 г., 20 сентября (9): 1347-52. [Ссылка QxMD MEDLINE].
webmd.com»> Wiggam MI, O’Kane MJ, Harper R, et al. Лечение диабетического кетоацидоза с нормализацией концентрации 3-гидроксибутирата в крови в качестве конечной точки неотложной помощи. Рандомизированное контролируемое исследование. Лечение диабета . 1997 г., 20 сентября (9): 1347-52. [Ссылка QxMD MEDLINE].Noyes KJ, Crofton P, Bath LE, et al. Тестирование гидроксибутирата рядом с пациентом для оценки новой конечной точки внутривенной инсулинотерапии при лечении диабетического кетоацидоза у детей. Педиатрический диабет . 2007 8 июня (3): 150-6. [Ссылка QxMD MEDLINE].
Фиордалиси И., Новотный В.Е., Холберт Д., Финберг Л., Харрис Г.Д. 18-летнее проспективное исследование диабетического кетоацидоза у детей: подход к минимизации риска грыжи головного мозга во время лечения. Педиатрический диабет . 2007 г. 8 июня (3): 142-9. [Ссылка QxMD MEDLINE].

Puttha R, Cooke D, Subbarayan A, Odeka E, Ariyawansa I, Bone M. Низкая доза (0,05 ЕД/кг/ч) сравнима со стандартной дозой (0,1 ЕД/кг/ч) внутривенной инфузии инсулина для начального лечения диабетического кетоацидоза у детей с сахарным диабетом 1 типа — обсервационное исследование. Педиатрический диабет . 6 июля 2009 г. [Ссылка QxMD MEDLINE].
Буткевич Э.К., Лейбсон К.Л., О’Брайен П.С., Палумбо П.Дж., Рицца Р.А. Инсулинотерапия при диабетическом кетоацидозе. Болюсная инъекция инсулина по сравнению с непрерывной инфузией инсулина. Лечение диабета . 1995 г., 18 августа (8): 1187-90. [Ссылка QxMD MEDLINE].
Della Manna T, Steinmetz L, Campos PR, Farhat SC, Schvartsman C, Kuperman H. Подкожное введение быстродействующего аналога инсулина: альтернативное лечение педиатрических пациентов с диабетическим кетоацидозом.
 Лечение диабета . 2005 г. 28 августа (8): 1856-61. [Ссылка QxMD MEDLINE].
Лечение диабета . 2005 г. 28 августа (8): 1856-61. [Ссылка QxMD MEDLINE].Грин С.М., Ротрок С.Г., Хо Д.Д. и др. Неспособность дополнительного бикарбоната улучшить исход при тяжелом педиатрическом диабетическом кетоацидозе. Энн Эмерг Мед . 1998 31 января (1): 41-8. [Ссылка QxMD MEDLINE].
Hale PJ, Crase J, Nattrass M. Метаболические эффекты бикарбоната при лечении диабетического кетоацидоза. Br Med J (Clin Res Ed) . 1984 г., 20 октября. 289(6451):1035-8. [Ссылка QxMD MEDLINE].
White H, Cook D, Venkatesh B. Использование гипертонического раствора для лечения внутричерепной гипертензии после черепно-мозговой травмы. Анест Анальг . 2006 июнь 102(6):1836-46. [Ссылка QxMD MEDLINE].
Ванелли М., Киари Г.
 , Гиззони Л. и др. Эффективность программы профилактики диабетического кетоацидоза у детей. 8-летнее обучение в школах и частных практиках. Лечение диабета . 1999 22 января (1): 7-9. [Ссылка QxMD MEDLINE]. [Полный текст].
, Гиззони Л. и др. Эффективность программы профилактики диабетического кетоацидоза у детей. 8-летнее обучение в школах и частных практиках. Лечение диабета . 1999 22 января (1): 7-9. [Ссылка QxMD MEDLINE]. [Полный текст].[Руководство] Миллер С.Г. Семейная терапия рецидивирующего диабетического кетоацидоза: рекомендации по лечению. Семейная системная медицина . 1996. 14(3):303-14.
Дуглас Д. Изотонические жидкости полезны при диабетическом кетоацидозе у детей: исследование. Медскейп. 4 апреля 2013 г. Доступно на http://www.medscape.com/viewarticle/781938. Доступ: 16 апреля 2013 г.
Белый ПК, Dickson BA. Низкая заболеваемость и смертность у детей с диабетическим кетоацидозом, получавших лечение изотоническими жидкостями. J Педиатр . 2013 г., 15 марта. [Ссылка на MEDLINE QxMD].

Детский диабетический кетоацидоз (ДКА)
Детский диабетический кетоацидоз (ДКА)
Диабетический кетоацидоз (ДКА) — это состояние, которое развивается, когда организм не может вырабатывать достаточное количество инсулина. Без инсулина глюкоза не может быть использована для получения энергии. Вместо этого тело будет использовать жир для получения энергии, в которой оно нуждается.
Что такое детский диабетический кетоацидоз (ДКА)?
Диабетический кетоацидоз (ДКА) возникает при повышенном уровне глюкозы в крови И кетонов в моче. Высокие кетоны — это кислоты, которые образуются, когда организм сжигает жир для получения энергии и когда не хватает инсулина.
Важно проверить мочу вашего ребенка, когда уровень глюкозы в крови равен или превышает 250 и когда ваш ребенок плохо себя чувствует или болен.
Что такое кетоны?
Кетоны — это молекулы кислоты, которые вырабатываются, когда организм голодает. Они могут быть опасны при диабете 1 типа и могут привести к диабетическому кетоацидозу, неотложной помощи при диабете.
Они могут быть опасны при диабете 1 типа и могут привести к диабетическому кетоацидозу, неотложной помощи при диабете.
Каковы признаки и симптомы детского диабетического кетоацидоза (ДКА)?
С симптомами, схожими с обычными заболеваниями, ДКА может быть трудно обнаружить, но конкретные подсказки могут помочь отличить их друг от друга. Вот почему важно знать признаки и симптомы ДКА, чтобы помочь распознать его на ранней стадии и обеспечить своевременную целенаправленную терапию.
Пять основных признаков и симптомов диабетического кетоацидоза включают
- Частое/учащенное мочеиспускание — Высокий уровень сахара в крови может привести к тому, что ваш ребенок мочится чаще, чем обычно. Проверьте образец мочи на глюкозу и кетоны, чтобы определить, присутствует ДКА или нет.
- Усиление жажды В ответ на частое мочеиспускание ваш ребенок может чувствовать обезвоживание и жажду сильнее, чем обычно.