
Болезнь кленового сиропа лейциноз: Болезнь кленового сиропа — что это?

Болезнь кленового сиропа — что это?
Причины болезни «кленового сиропа»
Данная болезнь является наследственной (аутосомно-рецессивный тип наследования). Частота заболеваемости – 1 чел. на 120-300 тыс. новорожденных.
Симптомы лейциноза
Первичный биохимический дефект заключен в резком снижении или полном отсутствии активности ферментной системы, отвечающей за окислительное декарбоксилирование аминокислот. Это приводит к накоплению аминокислот валины, лейцины и изолейцины в организме.
Первые проявления заболевания фиксируются в течение первых 2 недель после рождения ребенка. Типичные симптомы:
- ребенок отказывается от пищи;
- тихий плач;
- частые срыгивания, иногда – рвота.
При несвоевременном обращении к врачу болезнь прогрессирует: начинаются подергивания отдельных групп мышц, увеличение мышечного тонуса, ребенок вытягивается в теле, ноги при этом скрещиваются. Тяжелое течение болезни «кленового сиропа» может привести к нарушению дыхания. Если не лечить ребенка с первых недель жизни, прогноз на выздоровление чаще всего будет негативным. Ребенок будет сильно отставать в весе, в психологическом и физическом развитии.
Для диагностики заболевания достаточно наличия запаха мочи, напоминающего кленовый сироп. Присутствие аминокислот подтверждается с помощью клинических анализов.
Лечение болезни «кленового сиропа»
Для лечения лейциноза используют смеси аминокислот, а также белковые гидролизаты, которые освобождены от аминокислот с разветвленной цепью. Цель лечения – снизить уровень лейцина, изолейцина и валина, содержащихся в плазме крови. Часто для этой цели назначают специальную диету: ребенку дают микстуру из 18 аминокислот в пропорции, схожей с составом материнского молока. Вместо жиров в микстуре используется кукурузное масло, вместо углеводов — декстрин-мальтозой. В составе также есть витамины и минеральные вещества.
После улучшения общего состояния ребенка (а на это укажет нормальный аппетит, хороший мышечный тонус), в рацион вводят немного коровьего молока. Ребенок должен питаться согласно требованиям специальной диеты еще длительный период. Например, с 3 месяцев нужно вводить в рацион овощи и фрукты, с 6 месяцев – желатин.
Ребенок должен питаться согласно требованиям специальной диеты еще длительный период. Например, с 3 месяцев нужно вводить в рацион овощи и фрукты, с 6 месяцев – желатин.
Для диагностики и лечения болезни «кленового сиропа» рекомендуется обратиться к педиатру или генетику. Найти лучшего специалиста в своем городе и записаться к нему на прием можно, воспользовавшись возможностями сайта Doc.ua.
БОЛЕЗНЬ С ЗАПАХОМ КЛЕНОВОГО СИРОПА МОЧИ
БОЛЕЗНЬ С ЗАПАХОМ КЛЕНОВОГО СИРОПА МОЧИ
(ЛЕЙЦИНОЗ; КОРОТКО-ЦЕПОЧЕЧНАЯ КЕТОАЦИДУРИЯ )
MSUD; BRANCHED-CHAIN KETOACIDURIA; BRANCHED-CHAIN ALPHA-KETO ACID DEHYDROGENASE DEFICIENCY; BCKD DEFICIENCY; KETO ACID DECARBOXYLASE DEFICIENCY; MAPLE SYRUP URINE DISEASE, CLASSIC, INCLUDED; MAPLE SYRUP URINE DISEASE, INTERMEDIATE, INCLUDED;MAPLE SYRUP URINE DISEASE, INTERMITTENT, INCLUDED; MAPLE SYRUP URINE DISEASE, THIAMINE-RESPONSIVE, INCLUDED; MAPLE SYRUP URINE DISEASE, E3-DEFICIENT, WITH LACTIC ACIDOSIS, INCLUDED; MAPLE SYRUP URINE DISEASE, TYPE IA, INCLUDED; MSUD1A, INCLUDED; MAPLE SYRUP URINE DISEASE, TYPE IB, INCLUDED; MSUD1B, INCLUDED; MAPLE SYRUP URINE DISEASE, TYPE II, INCLUDED; MSUD2, INCLUDED; MAPLE SYRUP URINE DISEASE, TYPE III, INCLUDED; MSUD3, INCLUDED; LIPOAMIDE DEHYDROGENASE DEFICIENCY, LACTIC ACIDOSIS, CONGENITAL INFANTILE, DUE TO LAD DEFICIENCY, NCLUDED; DIHYDROLIPOAMIDE DEHYDROGENASE DEFICIENCY, INCLUDED DLD DEFICIENCY, INCLUDED
MIM#248600
Генетика: Генетически гетерогенное заболевание, которое связано с недостаточностью ферментного комплекса дегидрогеназ *-кетокислот с боковыми цепями (BCKAD). В состав BCKAD входит 4 субъединицы (E1a, E1b, E2, и E3). Мутации в 3 генах, кодирующие эти белки приводят к нарушению расщепления разветвленных аминокислот и накоплению соответствующих разветвленных органических кетокислот в биологических жидкостях и тканях. Ген, кодирующий E1a субъединицу (BCKDHA) картирован на 19q13.1-q13.2), субъединицу E1b (BCKDHB) картирован на 6q14), E2 (DBT) (картирован на 1p31), E3 (DLD) картирован на 7q31-q32. Мутации в гене E3 (DLD) приводят к заболеванию по клинической картине, сходному с синдромом Ли.
Мутации в гене E3 (DLD) приводят к заболеванию по клинической картине, сходному с синдромом Ли.
Тип наследования: аутосомно-рецессивный
Эпидемиология:Средняя частота 1:185 000 живых новорожденных. Заболевание распространено в высокоинбредных популяциях Пенсильвании — частота 1:380 живых новорожденных.
Патогенез: : Недостаточность ферментного комплекса дегидрогеназ *-кетокислот с боковыми цепями, сопровождающаяся накоплением кетокислот (превалирует *-кетоизокапроновая кислота) в биологических жидкостях и тканях, ведет к метаболическому кетоацидозу, гипераммониемии, токсически действует на центральную нервную систему, вызывая генерализованный или локальный (белое вещество мозжечка, ствол) отек мозга, гипомиелинизацию, атрофию. Безусловное нейротоксическое действие имеют лейцин и его кетокислота, нейротоксический эффект валина и его кетокислоты сомнителен. Вторично нарушаются другие метаболические процессы: снижается доступность субстратов для глюконеогенеза в печени — аланина и глютамина, избыточно потребляемых для реанимирования *-кетокислот, что ведет к гипогликемии. Запах «кленового сиропа» обусловлен накоплением *-кето-*-метилвалериановой кислоты.
Клинические проявления: Классическая (острая) форма наблюдается наиболее часто. Заболевание дебютирует с первой недели жизни с отказа от еды, необъяснимых рвот, судорог, летаргии, быстропрогрессирующей в кому. Развивается метаболический кетоацидоз и гипогликемия. Характерны потеря веса и прогрессирующая неврологическая симптоматика в виде нарушения мышечного тонуса, патологический рефлекс Моро, стереотипные движения по типу «педалирующих» или «боксирующих».
При промежуточной форме, заболевание может дебютировать с внезапно возникшей преходящей атаксии. В большинстве случаев болезнь манифестирует от 5 месяцев до 7 лет. Внезапное ухудшение может быть связано с инфекционными заболеваниями или стрессом, что напоминает классическую форму заболевания. Моча, пот, серная пробка могут иметь специфичный сладкий запах. Во время метаболического криза наблюдается повышение уровня лейцина, изолейцина, валина в крови и их метаболитов в моче. В неонатальном периоде и между метаболическими кризами данные показатели могут иметь нормальные значения.
Моча, пот, серная пробка могут иметь специфичный сладкий запах. Во время метаболического криза наблюдается повышение уровня лейцина, изолейцина, валина в крови и их метаболитов в моче. В неонатальном периоде и между метаболическими кризами данные показатели могут иметь нормальные значения.
Для интермиттирующей фоpмы характерно волнообразное течение. Заболевание манифестирует в возрасте от 5 месяцев до 2 лет, но иногда значительно позже. Приступы метаболического кетоацидоза, провоцируются факторами: вакцинацией, интеркуррентными инфекциями, высокобелковой диетой, оперативными вмешательствами. Заболевание манифестирует приступами pвот, дегидратации, прогрессирующей летаргией, судорогами, мозжечковой атаксией. В межприступный период пациенты не предъявляют жалоб и биохимические показатели (уровень аминокислот и органических кислот) может быть в пределах нормы. На высоте приступа метаболического кетоацидоза может развиться ступор или кома с летальным исходом.
Диагностика: При МРТ визуализируют признаки отека вещества головного мозга. Основными методами подтверждения диагноза являются биохимические. С помощью методов высокоэффективной жидкостной хроматографии или тандемной масс спектрометрии выявляют повышение концентрации аминокислот лейцина, изолейцина, валина в крови и моче. Методами газовой хроматографии выявляют повышение уровня соответсвующих кетокислот *-кетоизовалериановой, *-кето-*-метилвалеpиановой, *-кетоизокапроновой. Патогономоничные для заболевания биохимические маркеры — L-аллоизолейцин и *-гидрокси-изовалериановая кислота — производные накапливающихся продуктов — L-изолейцина и *-кетоизовалериановой кислоты.
При промежуточной форме концентрации аминокислот и кетокислот с боковыми цепями в биологических жидкостях увеличены в меньшей степени, чем при классической форме. Пpи интеpмиттиpующей фоpме — аминокислоты и кетокислоты с боковыми цепями повышены в крови и моче только во время кризов. Возможно проведение ДНК-диагностики. Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru) , Московском научно-исследовательском институте педиатрии и детской хирургии (http://www.pedklin.ru )
Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru) , Московском научно-исследовательском институте педиатрии и детской хирургии (http://www.pedklin.ru )
Лечение: Всем больным показано назначение гиперкалорийной диета с огpаничением лейцина, изолейцина, валина до минимальных уровней, необходимых для поддержания нормального роста и развития. Разработано несколько коммерчески доступных формул с дозированным содержанием аминокислот, жиpов, углеводов, калорийности, микроэлементов и витаминов, обеспечивающих хороший клинический эффект для больных разного возраста.
Лечение острых кризов. Для удаления токсических продуктов используют перитонеальный диализ и гемодиализ. Для поддержания питания больных в острой фазе разработана специальная формула для парентеральной терапии, не содержащая аминокислот с боковыми цепями. Для минимизации катаболических процессов используют инсулин подкожно в дозе 0,3 — 0,4 МЕ/кг/24 час на фоне введения углеводов в дозе 23 — 26 г/кг, соответственно. Тиамин назначают в дозах от 50 до 300 мг/день при постановке диагноза всем пациентам. Один из эффективных методов лечения заболевания — трансплантация печени, которая позволяет избежать строгих диетарных ограничений и предотвращает развитие метаболических кризов.
Прогноз
При ранней диагностике и тщательном метаболическом контроле прогноз заболевания относительно благоприятный.
Лейциноз (кленовый запах мочи) — МЦ Биосс, медицинский центр клиника БИОСС: 1 мин — метро Беговая, 10 мин
Кленовый запах мочи — явный симптом заболевания. Ниже в статье вы найдете причины заболевания; врачей, которые его лечат; необходимые лечебные процедуры для лечения; а также общую информацию о заболевании его локализации, особенностях диагностики заболеваний и лечения их. Тем не менее, советуем проконсультироваться с врачом, ибо самолечение в 90% чревато осложнениями
Запись на прием и консультацию
Лейциноз.
 Общая информация
Общая информация
Заболевание лейциноз называют еще «болезнью кленового сиропа» из-за характерного запаха мочи. Данная проблема связана с нарушением обмена аминокислот. В результате, в моче обнаружено повышенное количество вещества лейцина.
Данное заболевание встречается крайне редко, его часто очень сложно диагностировать ввиду того, что недуг может протекать практически бессимптомно у детей грудного возраста.
Различают несколько вариантов лейциноза:
-
классический; -
периодический; -
промежуточный; -
тиаминзависимый.
При лечении Лейциноза врачи клиники БИОСС используют как проверенные временем, так и самые новые разработки и авторские методики.
В МЦ БИОСС работают лучшие врачи в Москве, имеющие большой опыт в лечении Лейциноза
Симптомы Лейциноза (болезни кленового сиропа)
Основные симптомы болезни кленового сиропа (которые проявляются почти сразу после рождения):
— плохой аппетит,
— рвота,
— обезвоживание,
— вялость,
— гипотония,
— судороги,
— гипогликемия,
— кетоацидоз,
— опистотонус,
— панкреатит,
— кома
— и разнообразные неврологические нарушения.
Диагностика Лейциноза (болезни кленового сиропа)
Напоминающий кленовый сироп своеобразный запах мочи больных, в которой обнаруживается присутствие аминокислот с разветвлённой цепью.
При проведении селективного скрининга моча дает положительную реакцию с 2,4-динитрофенилгидразином.
Лечение Лейциноза (болезни кленового сиропа)
При лечении используют смеси аминокислот и белковые гидролизаты, освобождённые от аминокислот с разветвлённой цепью.
Состоит в снижении уровня лейцина, изолейцина и валина в плазме крови, для чего применяется специальная диета, в которой вместо белка назначается особая микстура, состоящая из смеси 18 аминокислот в той пропорции, как и в женском молоке. Жиры в такой смеси присутствуют в виде кукурузного масла, а углеводы заменяются декстрин-мальтозой. Также добавляются минеральные вещества и витамины. Как только общее состояние ребенка улучшается, появляется аппетит, нормализуется мышечный тонус, в рацион вводится небольшое количество коровьего молока. Фрукты и овощи, которые также содержат белок, но в небольшом количестве, разрешается давать ребенку уже с 3 месяцев. В 6-8 месяцев в рацион вводят желатин, который не содержит указанных аминокислот. С 9,5 месяцев ежедневная порция молока составляет 120 мл.
Что провоцирует / Причины Лейциноза (болезни кленового сиропа)
Наследственное заболевание с аутосомно-рецессивным типом наследования встречается с частотой 1 на 120-300 тыс. новорождённых.
Патогенез (что происходит?) во время Лейциноза (болезни кленового сиропа)
Первичный биохимический дефект заключается в отсутствии или резком снижении активности ферментной системы, обеспечивающей окислительное декарбоксилирование трёх аминокислот — лейцина, изолейцина и валина. В результате в организме накапливаются эти аминокислоты и их предшественники. Наиболее патогенно накопление лейцина.
«Живой» поиск
Вывести список по локализации заболеваний
Вывести полный алфавитный список заболеваний
Перейти к поиску по симптомам
Лейциноз, описание заболевания на портале Medihost.ru
Лейциноз относится к наследственным заболеваниям, связанным с нарушением аминикислотного обмена. Второе название этой патологии – болезнь кленового сиропа, поскольку у больных запах мочи очень схож с запахом этого вещества. При этом заболевании организм ребенка не может усваивать некоторые аминокислоты, а специфический запах в моче образуется за счет присутствия вещества, которое образуется из лейцина.
Причины лейциноза
Это заболевание вызывается снижением или полным отсутствием активности фермента декарбоксилазы альфа-кетокислот в организме ребенка. Поэтому аминокислоты не перерабатываются, а постепенно скапливаются в крови и моче ребенка, что оказывает негативное токсическое воздействие на его состояние.
Симптомы лейциноза
Симптоматика заболевания проявляется через несколько дней, после рождения ребенка. Ребенок отказывается от кормления и монотонно плачет. При попытках накормить малыша возникает рвота или срыгивание.
Основной симптом лейциноза – специфический запах мочи, который походит на запах жженого сахара или кленового сиропа.
Существует несколько форм заболевания, со своими характерными признаками:
- Острая форма. Проявляется на первых днях жизни ребенка. Симптоматика нарастает стремительно.
- Промежуточная форма. Проявляется от пяти месяцев до семи лет, в виде кратковременных расстройств координации движения и специфическим запахом мочи. Провоцируются эти приступы стрессовыми ситуациями, нарушением диеты, респираторными заболеваниями. В период ремиссии в развитии ребенка отклонений от нормы, как правило, не отмечается.
- Прерывающаяся форма. Имеет волнообразное течение, первые симптомы отмечаются с пятимесячного возраста и могут продолжаться до двух лет. Приступ может вызывать любой стрессовый фактор для организма ребенка: вакцинация, простуды, нарушение питания. Симптомы приступа выражаются в нарушении координации движения, выраженной рвоте, судорогах. При отсутствии своевременной помощи приступ может перетечь в кому, с дальнейшим летальным исходом. В период ремиссии состояние ребенка находится в пределах нормы.
Вырастают такие дети с задержкой физического и психомоторного развития.
Диагностика лейциноза
Для диагностирования этого заболевания проводится анализ анамнеза, сбор жалоб родителей и поведения ребенка. Оцениваются изменения в нем.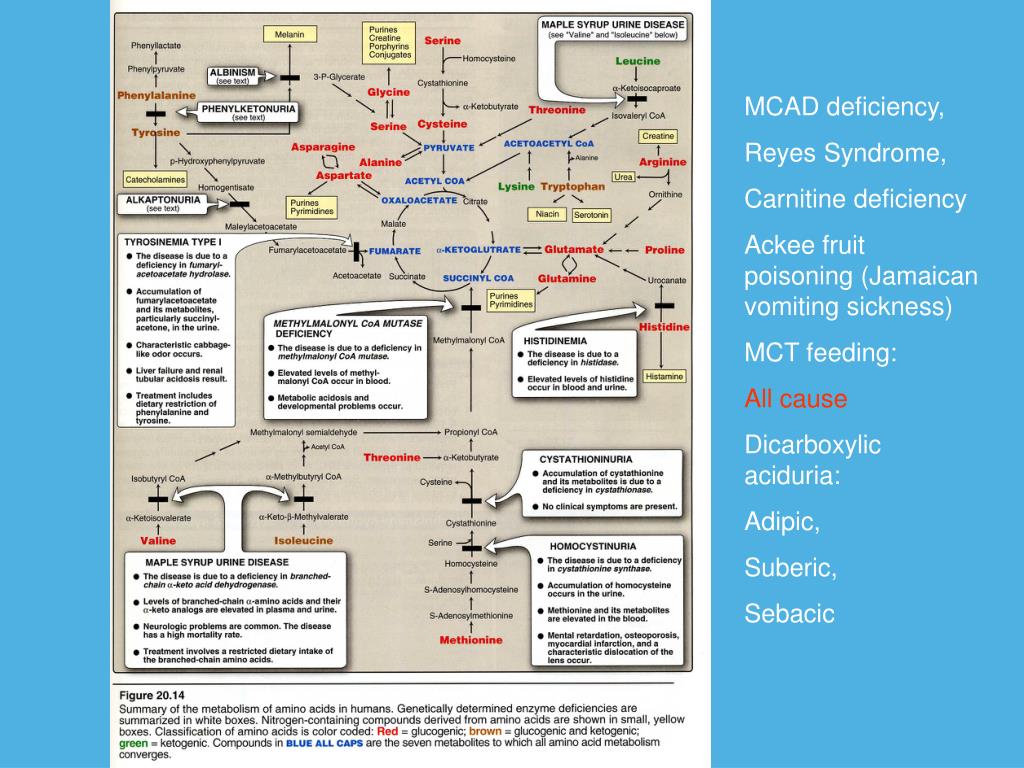 Осуществляется физикальный осмотр ребенка, взвешивание и определения мышечного тонуса.
Осуществляется физикальный осмотр ребенка, взвешивание и определения мышечного тонуса.
Затем проводятся лабораторные исследования для определения наличия в моче нерасщепленных аминокислот.
Кровь также исследуется на наличие нерасщепленных аминокислот, забор материала происходит как из пальца, так и из вены, на специализированной бумаге. С помощью этого метода можно своевременно обнаружить повышение в крови аминокислот, чтобы не допустить ухудшение состояния ребенка.
При необходимости назначаются дополнительные инструментальные исследования для определения общего состояния ребенка.
Лечение лейциноза
Основное направление терапии этого заболевания состоит в снижении уровня лейцина, изолейцина и валина в крови ребенка. Для этого назначается специальная диета, где белок заменен на особую микстуру, с тем же набором аминокислот, что и в материнском молоке. Назначается витаминотерапия и прием минералов. При улучшении состояния ребенка, когда появляется аппетит и нормализуется его мышечный тонус вводится минимальное количество коровьего молока, а также фрукты и овощи, содержащие белок.
К сожалению, даже соблюдение коррекционной диеты не гарантирует нормального психомоторного развития ребенка и набор веса. Общий прогноз этого заболевания неблагоприятный.
причины, симптомы, диагностика и лечение
Лейциноз – наследственная болезнь, в основе которой лежит дефицит дегидрогеназ кетокислот с разветвленной цепью, а также нарушение метаболизма аминокислот лейцина, изолейцина и валина. Проявляется патологической слабостью, тихим плачем, рвотой, мышечными подергиваниями, характерным «кленовым» запахом мочи, задержкой психомоторного развития. При кризе возникают нарушения дыхания и кровообращения, потеря сознания. Диагностика включает анализ на аминокислоты и производные (кровь, моча), молекулярное-генетическое исследование и скрининговые пробы мочи. Для лечения используется диета с протеиновыми смесями, освобожденными от разветвленно-цепочечных аминокислот (BCAA)./blood-and-urine-samples-with-medical-results-551987461-59a5a5a322fa3a00109171e7.jpg)
Общие сведения
Синонимы лейциноза – болезнь разветвленных кислот, разветвленноцепочечная кетонурия, короткоцепочечная кетоацидурия, болезнь мочи с запахом кленового сиропа, болезнь кленового сиропа. Происхождение синонимичных названий связано с историей исследования данной патологии. В начале 1954 года австрийско-американский детский невролог Дж. Х. Менкес впервые описал заболевание, при котором у больных определялся специфический запах мочи, схожий с запахом древесного сиропа и жженого сахара. Еще одно, менее распространенное название – синдром Менкеса. Болезнь отнесена к категории редких (орфанных, «сиротских») патологий. Диагностируется с частотой 1 случай на 120-300 тысяч новорожденных.
Лейциноз
Причины лейциноза
Развитие болезни обусловлено наличием пары мутационных генов, кодирующих производство дегидрогеназы разветвленных альфа-кетокислот. При недостаточности этого фермента нарушается процесс катаболического распада лейцина, аллоизолейцина, изолейцина, валина. Структура дегидрогеназы представлена четырьмя белковыми соединениями: Е3, Е2, альфа-субъединицей E1, бета-субъединицей Е1. Выявлена локализация генов, ответственных за синтез компонентов энзимных комплексов: a-субъединицу протеина E1 кодирует ген BCKDHA на 19 хромосоме, b-субъединицу E1 – ген BCKDHB на 6 хромосоме, Е2-протеин – ген DBT на 1 хромосоме, протеин Е3 – ген DLD на 7 хромосоме. Наследственная передача происходит аутосомно-рецессивно, заболевание проявляется у ребенка, получившего пару мутантных генов одной локализации. При передаче мутации только от одного родителя симптоматика отсутствует, потому что доминантный ген обеспечивает организм необходимым количеством фермента.
Патогенез
В основе заболевания лежит генетическая недостаточность дегидрогеназ кетокислот, имеющих боковые цепи. В жидкостях организма накапливаются разветвленно-цепочечные аминокислоты и их производные: 2-кетоизокапроновая, 2-кето-3-метилвалериановая и 2-кетоизвалериановая кислоты. Избыток лейцина и его метаболитов оказывает нейротоксическое действие. При этом возникает дефицит других аминокислот, таких как аланин, глицин, глутамин, тирозин, триптофан. Нарушается их транспорт внутри клеток, формируются расстройства нейромедиаторной передачи. Развивается кетоацидоз, гипогликемия на фоне снижения глюконеогенеза, гипонатриемия, гипераммониемия, угнетаются процессы окислительного фосфорилирования.
Избыток лейцина и его метаболитов оказывает нейротоксическое действие. При этом возникает дефицит других аминокислот, таких как аланин, глицин, глутамин, тирозин, триптофан. Нарушается их транспорт внутри клеток, формируются расстройства нейромедиаторной передачи. Развивается кетоацидоз, гипогликемия на фоне снижения глюконеогенеза, гипонатриемия, гипераммониемия, угнетаются процессы окислительного фосфорилирования.
При форме лейциноза, вызванной деструкцией протеина E3, патогенез более сложен. Данная субъединица является компонентом нескольких ферментных комплексов, которые принимают участие в метаболизме пирувата и осуществлении цикла Кребса. В результате структурного и функционального дефекта этих комплексов меняется энергетический обмен клеток, возникает лактатацидоз.
Классификация
На начальных этапах исследования лейциноз описывался как быстро прогрессирующее, развивающееся с первых дней жизни и неизбежно приводящее к летальному исходу заболевание. Позже было установлено, что существует несколько разновидностей патологии с разным временем дебюта, течением и прогнозом. В настоящее время выделяют пять форм лейциноза:
- Острая. Также называется классической. Проявляется с рождения, симптомы нарастают стремительно даже при своевременном лечении. Активность энзима составляет менее 2% от нормы.
- Промежуточная. Нередко дебютирует в раннем и дошкольном детстве. Имеет приступообразное течение, ведущим симптомом является атаксия. Приступы провоцируются острыми заболеваниями, стрессом, отклонением от диеты. Фермент выполняет свои функции на 3-30%.
- Интермиттирующая. Другие названия – перемежающаяся, прерывающаяся. Обычно диагностируется в возрасте 5-24 месяцев. Приступы протекают остро с судорогами, дегидратацией, атаксией. При отсутствии неотложной помощи возникает кома. Ферментативная активность колеблется от 5 до 20%.
- Тиаминзависимая.
 Развивается в неонатальном периоде. По характеру течения может напоминать промежуточную или интермиттирующую формы. В ремиссии признаки болезни отсутствуют. Энзим активен на 30-40%.
Развивается в неонатальном периоде. По характеру течения может напоминать промежуточную или интермиттирующую формы. В ремиссии признаки болезни отсутствуют. Энзим активен на 30-40%. - E3-дефицитная. Определяется у младенцев со 2 месяца жизни. Сопровождается лактатацидозом, нарушением энергетического обмена. Относится к группе митохондриальных патологий.
Симптомы лейциноза
У детей с классической формой заболевания симптоматика появляется в первую неделю жизни. Начало всегда острое. Дебют лейциноза характеризуется резким ухудшением общего состояния новорожденного – развитием генерализованных судорог, патологической возбудимости, гипертонии мышц, неукротимой рвоты, обезвоженности. Ребенок кричит, отказывается есть. Состояние возбужденности сменяется патологической вялостью. Обнаруживаются признаки угнетения ЦНС, нарастает сомноленция, а затем кома. Поверхность кожи слущивается, покрывается эритемами. Моча пахнет кленовым сиропом. Ребенок чрезмерно уязвим в отношении инфекций, отстает в психическом, моторном развитии. Заболевание завершается скорой смертью, основной причиной которой становится отек головного мозга.
При интермиттирующей форме приступы сопровождаются возникновением мозжечковой атаксии – у больного нарушается походка и координация движений. Между приступами клиническая симптоматика отсутствует. Промежуточный лейциноз протекает тяжелее. Приступы не имеют четких временных границ, волнообразно нарастают и затихают. Дети отстают в развитии, страдают от атаксии, судорог и мышечной гипотонии, поздно осваивают ходьбу, манипулятивные действия, речь. Симптомы тиаминзависимой формы могут быть легкими или умеренными. Как и двух предыдущих вариантах, доминируют признаки нарушения походки и координации движений, мышечный тонус снижен, возможны судороги. У пациентов с Е3-зависимой формой наблюдается прогрессирующее отставание моторного и психического развития, рвота, мышечный гипотонус, расстройства дыхания. Периодически выявляется гипогликемия с апатией, слабостью, головокружениями, повышенной сонливостью.
Периодически выявляется гипогликемия с апатией, слабостью, головокружениями, повышенной сонливостью.
Осложнения
При совместимых с жизнью формах лейциноза основным осложнением является отставание в психическом и физическом развитии. Выраженность нарушений варьируется: иногда больные способны к самообслуживанию, ведут автономный образ жизни, посещают школу, в других случаях нуждаются в постоянном уходе, с трудом осваивают простые двигательные навыки и речь. Отсутствие медицинской помощи пациентам с острой формой лейциноза и при кризах приводит к тяжелым осложнениям – отеку мозга, острой недостаточности почек и печени. Данные состояния становятся причиной смерти больных.
Диагностика
Обследованию на лейциноз подлежат все дети с отягощенным семейным анамнезом, особенно если диагноз был подтвержден у старших братьев и сестер, а также если у новорожденного после периода удовлетворительного состояния проявились характерные симптомы: отказ от еды, рвота, судороги, патологическое возбуждение или летаргия, атаксия, кома, ацидоз. Диагностика осуществляется специалистами разных направлений: педиатрами, неврологами, врачами-генетиками. На начальном этапе проводится изучение анамнеза и жалоб родителей, общий осмотр ребенка с определением мышечного тонуса и веса, оценкой двигательной активности. С целью подтверждения диагноза, исключения гипоксических поражений ЦНС, внутримозговых кровоизлияний, острых инфекций и наследственных патологий обмена веществ назначаются лабораторные исследования:
- Аминокислотный анализ крови. Выявляется повышение уровня BCAA в крови, моче. Высокоспецифический маркер заболевания – увеличение содержания аллоизолейцина в плазме крови, показатель находится в диапазоне от 72 до 220 мкмоль/л. Концентрация изолейцина составляет 200-12 000 мкмоль/л, лейцина – 500-5 000 мкмоль/л, валина – 500-1 800 мкмоль/л.
- Анализ аминокислот, кетокислот мочи. Обнаруживается повышение показателей аминокислот в моче.
 Увеличено количество разветвленных кетокислот (в ммоль/ммоль креатинина): концентрация 2гидрокси-3-метилвалериановой кислоты – 60-400, 2гидрокси-изокапроновой – 3-80, 2гидрокси-изовалериановой – 850-3 600, 2кето-3метилвалериановой – 500-2 500, 2-кето-изокапроновой – 400-4 400, 2-кето-изовалериановой – 300-800.
Увеличено количество разветвленных кетокислот (в ммоль/ммоль креатинина): концентрация 2гидрокси-3-метилвалериановой кислоты – 60-400, 2гидрокси-изокапроновой – 3-80, 2гидрокси-изовалериановой – 850-3 600, 2кето-3метилвалериановой – 500-2 500, 2-кето-изокапроновой – 400-4 400, 2-кето-изовалериановой – 300-800. - Специфические тесты. Для экспресс-диагностики повышения уровня метаболитов аминокислот проводится реакция с 2,4-динитрофенилгидразином и проба Фелинга. Биоматериал – порция разовой мочи. При лейцинозе оба теста дают резко положительный результат. Поскольку методы обладают низкой чувствительностью и специфичностью, их применение целесообразно только в рамках скрининга.
- Молекулярно-генетический анализ. Подтверждение первичного дефекта необходимо при пренатальной и преимплантационной диагностике, при обследовании новорожденных не является обязательным. У больных выявляются мутации гомозиготного и компаунд-гетерозиготного сочетания в генах BCKDHA, BCKDHB, DBT и DLD (типы 1А, 1B, 2 и 3 соответственно).
Лечение лейциноза
Терапевтическая тактика ориентирована на подавление производства токсичных метаболитов BCAA, стимуляцию процессов анаболизма, снижение скорости кетоацидоза, предупреждение отека мозга и поражения внутренних органов. Лечебные мероприятия всем больным проводятся в плановом режиме, при развитии острого криза – в экстренном порядке. Плановые процедуры включают:
- Специальную диету. Система питания разрабатывается для каждого ребенка, но базируется на общих принципах. Белковая пища заменяется белковыми гидролизатами, не содержащими валина, изолейцина, лейцина. Источником жиров служит кукурузное масло, источником углеводов – декстринмальтоза. Дополнительно в рацион вводятся минеральные вещества и витамины. Контролируется энергетическая ценность питания, позволяющая поддерживать процессы анаболизма.
- Кофакторную терапию.
 При подтверждении диагноза лейциноз и до установления его формы осуществляется пробная терапия тиамином. Если лечение, проводимое на протяжении двух недель, нормализует концентрацию аминокислот с разветвленной боковой цепью, делается вывод о тиаминзависимой форме болезни. Прием тиамина продолжают в минимально эффективной дозировке.
При подтверждении диагноза лейциноз и до установления его формы осуществляется пробная терапия тиамином. Если лечение, проводимое на протяжении двух недель, нормализует концентрацию аминокислот с разветвленной боковой цепью, делается вывод о тиаминзависимой форме болезни. Прием тиамина продолжают в минимально эффективной дозировке. - Терапию левокарнитином. Вспомогательным методом этиотропной терапии является применение левокарнитина. Препарат усиливает связывание аминокислотных метаболитов, снижая уровень их свободных форм. Лечение назначается на срок от 3 до 6 месяцев.
- Симптоматическую терапию. В дополнение к основной терапии пациентам показаны лекарственные средства, купирующие симптомы болезни. Подбор препаратов производится индивидуально с учетом особенностей клинической картины. Используются витамины группы B, антиконвульсанты, противорвотные, ноотропы.
Метаболический криз требует немедленной госпитализации и проведения интенсивной терапии. Больных полностью переводят на питание из смеси аминокислот. При отказе от пищи или частой рвоте используют зонд, гастростому. Высокая калорийность питания обеспечивается введением растворов глюкозы и мальтодекстрина. Токсические продукты метаболизма удаляют с помощью перитонеального диализа и гемодиализа. При отеке головного мозга вводят гипертонический солевой раствор, диуретики.
Прогноз и профилактика
Классическая (острая) форма лейциноза характеризуется тяжелым прогрессивным течением. Ее прогноз неблагоприятен, эффективная терапия не разработана, сохраняется высокий риск летального исхода. При других вариантах болезни строгое соблюдение диеты и назначение медикаментозной терапии позволяет повысить качество жизни больных, сократить отставание общего развития, улучшить социальную адаптацию. Профилактика заключается в проведении пренатальной диагностики – определении аминокислотного состава амниотической жидкости, молекулярно-генетическом исследовании полученного при биопсии хориона материала с выявлением мутаций соответствующих генов.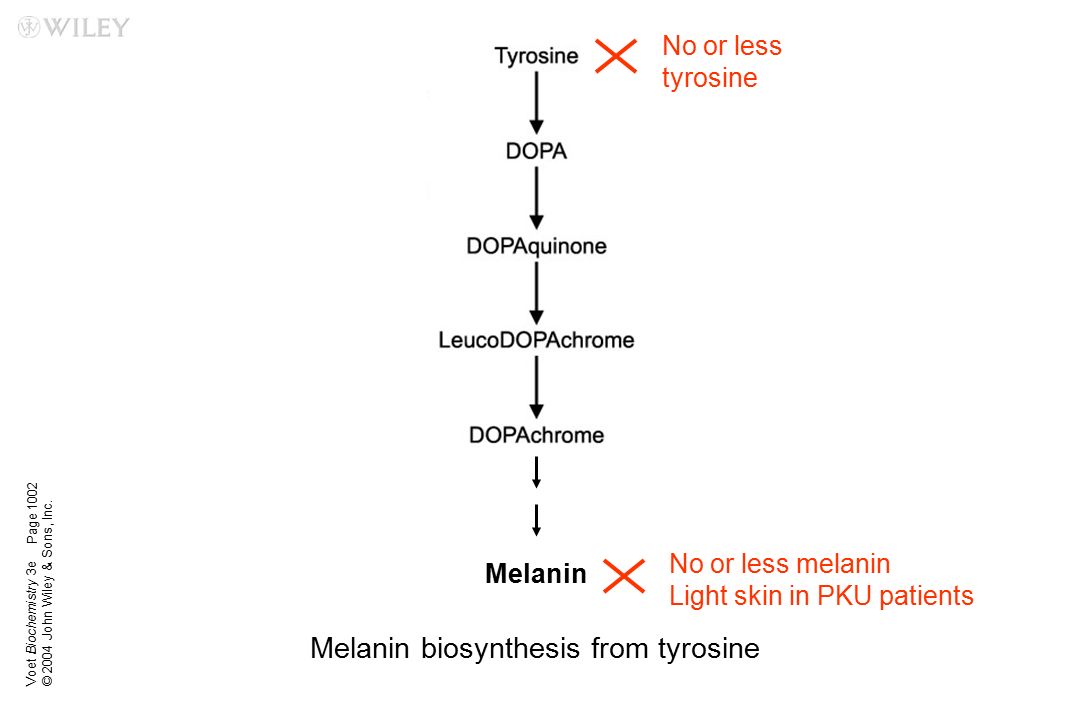
info — Азбука Genetics-info: Кленового сиропа болезнь
Болезнь кленового сиропа (лейцино́з, разветвлённоцепочечная кетонурия, болезнь мочи с запахом кленового сиропа) — наследственное заболевание из группы органических ацидемий, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма аминокислот лейцина, изолейцина, валина.
6292 •
•
15.04.2019
Впервые лейциноз был описан в 1954 г. доктором Менкесом. Он наблюдал ребенка с прогрессирующей детской церебральной дисфункцией. Заболевание манифестировало в первую неделю жизни, а в возрасте 3 месяцев ребенок умер. Внимание врачей привлек необычный симптом: моча ребенка имела запах, напоминающий запах кленового сиропа.
Болезнь кленового сиропа может быть вызвана гомозиготной мутацией или соединением гетерозиготных мутаций по крайней мере в 3 генах: BCKDHA на хромосоме 19q13, BCKDHB на хромосоме 6q14, и DBT на хромосоме 1р21. Эти гены кодируют 2 каталитических компонента комплекса альфа-кето-кислотных дегидрогеназ (BCKDC) с разветвленной цепью, который катализирует катаболизм аминокислот с разветвленной цепью, лейцина, изолейцина и валина. Мутация в третьем компоненте, E3 (DLD; 238331), на хромосоме 7q31, вызывает совмещенный, но более тяжелый фенотип, известный как дефицит DLD (DLDD; 246900).
Выделяют несколько клинико-генетических форм болезни кленового сиропа, для каждой из которых характерна своя картина:
Классическая, или неонатальная. Первые симптомы появляются на первой неделе жизни. Остро ухудшается общее состояние, отмечаются генерализованные судороги, повышенная возбудимость, резкий крик, отказ от пищи, рвота, мышечная гипертония, появляются признаки обезвоживания. Затем возбуждение сменяется вялостью, угнетением центральной нервной системы, сомноленцией, комой. Периоды мышечного гипертонуса чередуются с гипотонией. На коже появляются эритематозные высыпания, сухость. Появляется необычный запах мочи. Дети резко отстают в психомоторном развитии. Отмечается склонность к повторным инфекционным заболеваниям. МРТ-исследование головного мозга выявляет признаки диффузного отека мозговой ткани, изменение белого вещества мозга, мозжечка и базальных ганглиев, в частности, белого шара, таламуса, ствола мозга, внутренней и наружной капсулы.
На коже появляются эритематозные высыпания, сухость. Появляется необычный запах мочи. Дети резко отстают в психомоторном развитии. Отмечается склонность к повторным инфекционным заболеваниям. МРТ-исследование головного мозга выявляет признаки диффузного отека мозговой ткани, изменение белого вещества мозга, мозжечка и базальных ганглиев, в частности, белого шара, таламуса, ствола мозга, внутренней и наружной капсулы.
Промежуточная. Отличается менее злокачественным течением без выраженных приступов. Больные отстают в психомоторном развитии, у них отмечаются судороги, мышечная гипотония.
Интермиттирующая. Характеризуется выраженным приступообразным течением. В межприступном периоде у детей часто не обнаруживается никакой клинической симптоматики.
Тиамин-зависимая. При этом варианте заболевание обычно манифестирует после неонатального периода. Обменные расстройства в период метаболического криза характеризуются ацидозом, кетозом, высоким содержанием молочной кислоты в крови, гипогликемией. В крови и моче накапливаются лейцин, изолейцин, валин, а также аллоизолейцин — диагностический маркер заболевания. Моча имеет необычный запах. В крови снижено содержание аланина, глицина, может наблюдаться гипонатриемия.
Обусловленная дефицитом Е3-протеина и сопровождающаяся лактат-ацидозом (митохондриальное заболевание). Первые признаки обычно появляются в возрасте старше 8 недель. Характеризуется прогрессирующим отставанием психомоторного развития, рвотой, гипотонией/дистонией, нарушением дыхания. Выявляется метаболический ацидоз, повышение в крови уровня лактата, пирувата, α-кетоглутаровой кислоты, лейцина, изолейцина, валина, периодическая гипогликемия.
Диагностика болезни кленового сиропа основана на анализе родословной, оценке анамнеза, клинических проявлений, результатах анализа уровня аминокислот лейцина, изолейцина, валина в крови с подсчетом соотношения лейцин/аланин, определении почечной экскреции 2-кетоизокапроновой, 2-кето-3-метилвалериановой, 2- кетоизовалериановой кислот.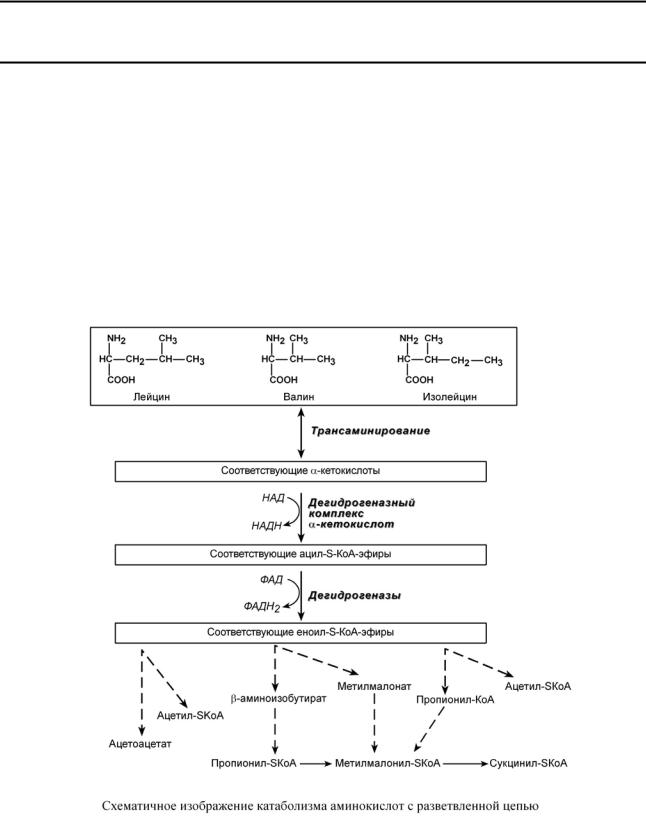 Основными методами подтверждения диагноза являются: тандемная масс-спектрометрия, аминокислотный анализ, газовая хроматография-масс-спектрометрия. Для подтверждения диагноза и медико-генетического консультирования проводится молекулярно-генетическое исследование.
Основными методами подтверждения диагноза являются: тандемная масс-спектрометрия, аминокислотный анализ, газовая хроматография-масс-спектрометрия. Для подтверждения диагноза и медико-генетического консультирования проводится молекулярно-генетическое исследование.
Относится к числу наследственных заболеваний, при которых диетотерапия является в настоящее время единственным методом лечения. В случае низкой эффективности проводимой терапии, при неблагоприятном течении болезни с частыми кетоацидотическими кризами осуществляют трансплантацию печени.
Прогноз заболевания относительно благоприятный при ранней диагностике и тщательном метаболическом контроле. Частота среди новорожденных 1:185000.
Лейциноз
Пользователи также искали:
лейциноз биохимия,
лейциноз диагностика,
лейциноз форум,
лейциноз генетика,
лейциноз клинические рекомендации,
лейциноз презентация,
лейциноз прогноз,
лейциноз у взрослых,
лейциноз,
Лейциноз,
лейциноз клинические рекомендации,
лейциноз презентация,
лейциноз у взрослых,
лейциноз диагностика,
лейциноз генетика,
лейциноз прогноз,
лейциноз биохимия,
прогноз,
биохимия,
клинические,
рекомендации,
презентация,
взрослых,
диагностика,
генетика,
форум,
лейциноз форум,
заболевания по алфавиту. лейциноз,
лейциноз,
…
Болезнь мочи с кленовым сиропом — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Danner DJ. Болезнь мочи кленового сиропа. Руководство NORD по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 468-9.
Scriver CR, Beaudet AL, Sly WS и др. Ред. Метаболические молекулярные основы наследственного заболевания. 8-е изд. Компании McGraw-Hill. Нью-Йорк, штат Нью-Йорк; 2001: 1971-96.
СТАТЬИ ИЗ ЖУРНАЛА
Simon E, Flaschker N, Schadewaldt P, Langenbeck U, Wendel U.Вариант болезни мочи кленового сиропа (МСУД) — весь спектр. J Inherit Metab Dis. 2006; 29: 716-24.
Chuang DT, Chuang JL, Wynn RM. Уроки генетических нарушений метаболизма аминокислот с разветвленной цепью. J Nutr. 2006; 136: 243С-9С.
Strauss KA, Mazariegos GV, Sindhi R, et al., Избирательная трансплантация печени для лечения классической болезни мочи кленовым сиропом. Am J Transplant. 2006; 6: 557-64.
Ogier de Baulny H, Saudubray JM. Органические ацидурии с разветвленной цепью.Semin Neonatal. 2002; 7: 65-74.
Saudubray JM, Nassogne MC, de Lonlay P, et al. Клинический подход к наследственным нарушениям обмена веществ у новорожденных: обзор. Semin Neonatal. 2002; 7: 3-15.
Мортон Д.Х., Штраус К.А., Робинсон Д.Л. и др. Диагностика и лечение болезни мочи кленовым сиропом: исследование 36 больных. Педиатрия. 2002; 109: 999-1008.
Wendel U, Saudubray JM, Bodner A, et al. Трансплантация печени при болезни мочи кленового сиропа. Eur J Pediatr. 1999; 158 Приложение 2: S60-64.
Рашед М.С., Рахбини З., Озанд П.Т. Применение тандемной масс-спектрометрии с электрораспылением для скрининга новорожденных. Семин Перинатол. 1999; 23: 183-93.
Chuang DT. Болезнь мочи кленовым сиропом: она прошла долгий путь. J Pediatr. 1998; 132: S17-23.
ИНТЕРНЕТ
Strauss KA, Puffenberger EG, Carson VJ. Болезнь мочи кленового сиропа. 30 января 2006 г. [Обновлено 23 апреля 2020 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2020 гг.Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1319/ По состоянию на 3 июня 2020 г.
Болезнь мочи кленового сиропа. 30 января 2006 г. [Обновлено 23 апреля 2020 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2020 гг.Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1319/ По состоянию на 3 июня 2020 г.
Defendi GL. Болезнь мочи кленового сиропа. Обновлено: 2 мая 2018 г. Доступно по адресу: http://www.emedicine.com/ped/topic1368.htm По состоянию на 3 июня 2020 г.
McKusick VA., Ed. Интернет-Менделирующее наследование в человеке (OMIM). Балтимор. MD: Университет Джона Хопкинса; Запись №: 248600; Последнее обновление: 12.07.2018. Доступно по адресу: http://omim.org/entry/248600. Проверено 3 июня 2020 г.
Болезнь мочи кленовым сиропом.Домашний справочник по генетике. Проверено в июле 2017 г. Доступно по адресу: http://ghr.nlm.nih.gov/condition/maple-syrup-urine-disease. По состоянию на 3 июня 2020 г.
Maple Syrup Urine Disease (MSUD)
Краткий обзор
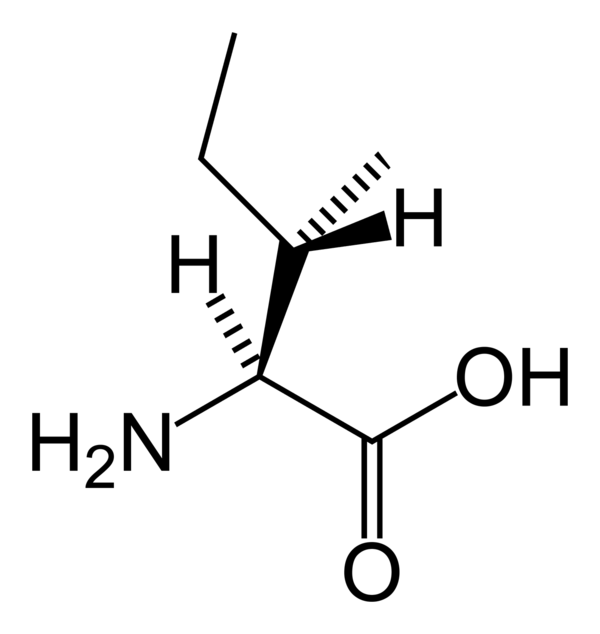
Болезнь мочи кленовым сиропом (MSUD) — это наследственное нарушение метаболизма незаменимых аминокислот лейцина, изолейцина и валина, вызванное дефектом комплекса дегидрогеназы альфа-кетокислот с разветвленной цепью. Эти три аминокислоты с разветвленной цепью (BCAA) присутствуют во всех белковых продуктах, и у людей с MSUD будут повышенные уровни всех трех.
MSUD наследуется по аутосомно-рецессивному типу. Аутосомно-рецессивное наследование означает, что человек унаследовал две аномальные копии гена (каждый ген содержит мутацию), вызывая MSUD. MSUD может быть вызван одним из трех разных генов: BCKDHA, BCKDHB и DBT. Оба родителя человека с MSUD являются носителями и не проявляют никаких симптомов заболевания.
MSUD может быть представлен в четырех различных формах. Классический MSUD часто проявляется в период новорожденности, обычно в течение первой недели жизни — даже уже через 2 дня жизни; MSUD среднего уровня может присутствовать в любом возрасте; прерывистый МСУД может проявляться только эпизодами атаксии и кетоацидоза, связанными с провоцирующими заболеваниями; и четвертая форма MSUD считается чувствительной к добавкам тиамина. У нелеченных пациентов обычно наблюдается специфический запах кленового сиропа (описан сладкий или карамельный) в моче или серной пробе из-за повышенного содержания 4,5-диметил-3-гидрокси-2 [5H] -фуранона (сотолона). У заболевших младенцев наблюдается энцефалопатия с кетоацидозом.
У нелеченных пациентов обычно наблюдается специфический запах кленового сиропа (описан сладкий или карамельный) в моче или серной пробе из-за повышенного содержания 4,5-диметил-3-гидрокси-2 [5H] -фуранона (сотолона). У заболевших младенцев наблюдается энцефалопатия с кетоацидозом.
У этого новорожденного может не быть очевидного триггера, но ухудшение симптомов неумолимо. Младенцы обладают пониженной реактивностью, раздражительностью и плохим кормлением и прогрессируют до комы. Дыхательная недостаточность, брадикардия, икота и переохлаждение появляются по мере увеличения отека головного мозга (часто называемого «лейцинозом»).Движения, описываемые как «фехтование» или «езда на велосипеде», могут возникать при апноэ и опистотонусе. Считается, что отек возникает из-за дисбаланса электролитов в результате накопления кетокислот с разветвленной цепью (BCKA) и аминокислот. В частности, считается, что лейцин и 2-кетоизокапроновая кислота (2 из BCKA) способствуют отеку мозга и токсичности. Считается, что валин и изолейцин и их соответствующие метаболиты кетокислот не вносят значительного вклада в токсичность.
Необходимо учитывать другие заболевания, которые могут вызывать острую энцефалопатию, такие как вирусный и бактериальный менингит, токсичность и передозировка лекарств, а также наследственные заболевания, вызывающие тяжелый метаболический ацидоз, лактоацидоз и / или гипераммониемию.
Какие тесты мне следует запросить, чтобы подтвердить мой клинический Dx? Кроме того, какие дополнительные тесты могут быть полезны?
MSUD диагностируется путем обнаружения повышенных уровней BCAA: лейцина, изолейцина и валина. Аллоизолейцин также обнаруживается у всех пациентов с тяжелыми и легкими формами, когда они не ограничивают потребление белка. Подъемы выявляются при стандартном количественном аминокислотном анализе плазмы. Аллоизолейцин также может совместно элюироваться с другими аминокислотами, чаще всего с изолейцином или цистатионином, что затрудняет обнаружение и количественную оценку. Если клинические подозрения высоки, следует связаться с директором лаборатории биохимической генетики для обсуждения и просмотра хроматограммы.
Если клинические подозрения высоки, следует связаться с директором лаборатории биохимической генетики для обсуждения и просмотра хроматограммы.
Дополнительные лабораторные исследования включают в себя органические добавки в моче, которые продемонстрируют повышение уровня производных 2-кето- и 2-гидрокси-BCKA. Прикроватное измерение содержания кетонов в моче может быть полезным для немедленного обнаружения повышенных кетокислот дома, в клинике или больнице, тем самым ускоряя быстрое лечение известных пациентов. Динитрофенилгидразин (DNPH) может быть добавлен в мочу и при наличии этих BCKA приведет к образованию желто-белого осадка.
Электролиты сыворотки, лактат, аммиак плазмы, ацилкарнитины, функция почек, общий анализ крови (CBC), функция печени и параметры свертывания должны быть проверены для оценки основного заболевания и в случае необходимости диализа.
Панели скрининга новорожденных теперь были расширены во всех штатах США и в большинстве развитых стран, и теперь они включают измерение BCAA. Обнаружение и количественное определение аллоизолейцина затруднено и возможно только в определенных центрах.Младенцу с повышенным уровнем лейцина / изолейцина и валина при скрининге новорожденных следует немедленно пройти клиническое обследование и провести анализ аминокислот в плазме.
Есть ли факторы, которые могут повлиять на результаты лабораторных исследований? В частности, принимает ли ваш пациент какие-либо лекарства — безрецептурные или травяные — которые могут повлиять на результаты лабораторных исследований?
Лица, у которых есть сомнительные результаты, должны пройти повторное тестирование после приема белка натощак или в течение первого дня жизни. Уровни аллоизолейцина менее 5 мкМ следует повторить и клинически согласовать с лечащим врачом.По возможности, образцы следует собирать во время острых симптомов, когда результаты неясны или клиническое описание противоречиво.
Повышение уровня BCAA без аллоизолейцина также может наблюдаться у лиц с тяжелым метаболическим кетоацидозом или длительным голоданием. Те, у кого есть подозрение на более легкие формы, все же должны иметь определяемый аллоизолейцин более 5 мкМ.
Те, у кого есть подозрение на более легкие формы, все же должны иметь определяемый аллоизолейцин более 5 мкМ.
Если скрининговый образец новорожденного или аминокислоты плазмы собираются до достаточного количества протеина, уровни BCAA не могут быть повышены.
Какие лабораторные результаты являются полностью подтверждающими?
Для диагностики достаточно
аминокислот в плазме с обнаружением повышенного уровня BCAA и аллоизолейцина. Также доступно дополнительное подтверждение с помощью секвенирования ДНК.
Хотя MSUD обычно встречается редко, с частотой около 1 на 180000 живорождений, мутации-основатели распространены в популяциях меннонитов в Пенсильвании, Огайо, Индиане, Мичигане, Кентукки, Висконсине и Нью-Йорке с частотой около 1 на 380 живорождений. рождений.Большинство мутаций уникальны, и сообщалось о сложных гетерозиготах с мутациями в разных генах.
Ферментный анализ можно провести в ткани печени, лейкоцитах или фибробластах кожи, но обычно в этом нет необходимости.
Какие тесты мне следует запросить, чтобы подтвердить мой клинический Dx? Кроме того, какие дополнительные тесты могут быть полезны?
Быстрое начало лечения при постановке диагноза, а также во время любого острого события имеет решающее значение. Повышение уровня лейцина и 2-кетоизокапроновой кислоты специфически нейротоксично, и повреждение головного мозга прогрессирует.Пострадавших помещают на диету с низким содержанием белка BCAA и строгим контролем за потреблением лейцина. Мониторинг с регулярным измерением аминокислот, электролитов, минералов, питательных микроэлементов, преальбумина, альбумина, железа, гемоглобина и незаменимых жирных кислот в плазме имеет решающее значение для обеспечения надлежащего клинического роста и развития. Недостатки следует немедленно устранять. Постоянное клиническое ведение с тщательным мониторингом диетического белка важно для обеспечения адекватного роста и развития.
Лечение во время острых событий включает прекращение приема белка и, возможно, гемодиализ для снижения накопления лейцина и кетокислот.Меры предосторожности по контролю внутричерепного давления с частыми неврологическими обследованиями и мониторингом отека мозга являются обязательными. За адекватной регидратацией с помощью болюсного раствора физиологического раствора следует вливание небелковых калорий, таких как внутривенные растворы 10% декстрозы, внутривенные интралипиды и назогастральное кормление смесью без BCAA. Внутривенное введение инсулина также может быть использовано для улучшения состояния анаболического метаболизма. Добавки с растворами изолейцина и валина также могут способствовать синтезу белка и снижению уровня лейцина.
Долговременная заболеваемость может включать остеопороз, а рецидивирующие инфекции Candida могут возникать из-за неправильного питания. В других сообщениях показано учащение случаев острого панкреатита, которое может быть трудно обнаружить у остро больного человека, страдающего рвотой.
Помимо классической тяжелой формы существуют другие формы, обусловленные остаточной ферментативной активностью. Средний MSUD может проявляться в более позднем возрасте с задержкой в развитии, плохим кормлением и увеличением веса. Эти люди все еще подвержены риску серьезных неврологических осложнений из-за отека мозга, когда они находятся в состоянии сильного стресса и катаболизма.Управление и наблюдение за этими людьми такое же, как и в классической форме.
Более легкий прерывистый MSUD может также сопровождаться тяжелым острым лейцинозом, но в анамнезе будет иметь нормальное развитие и рост. У них может быть только небольшое повышение уровня ВСАА, когда они здоровы, но при катаболическом стрессе будут проявляться более классические симптомы и лабораторные отклонения.
Описана форма MSUD, чувствительная к тиамину. Тиамин является кофактором ферментного комплекса и считается полезным для людей с более высоким уровнем остаточных ферментов.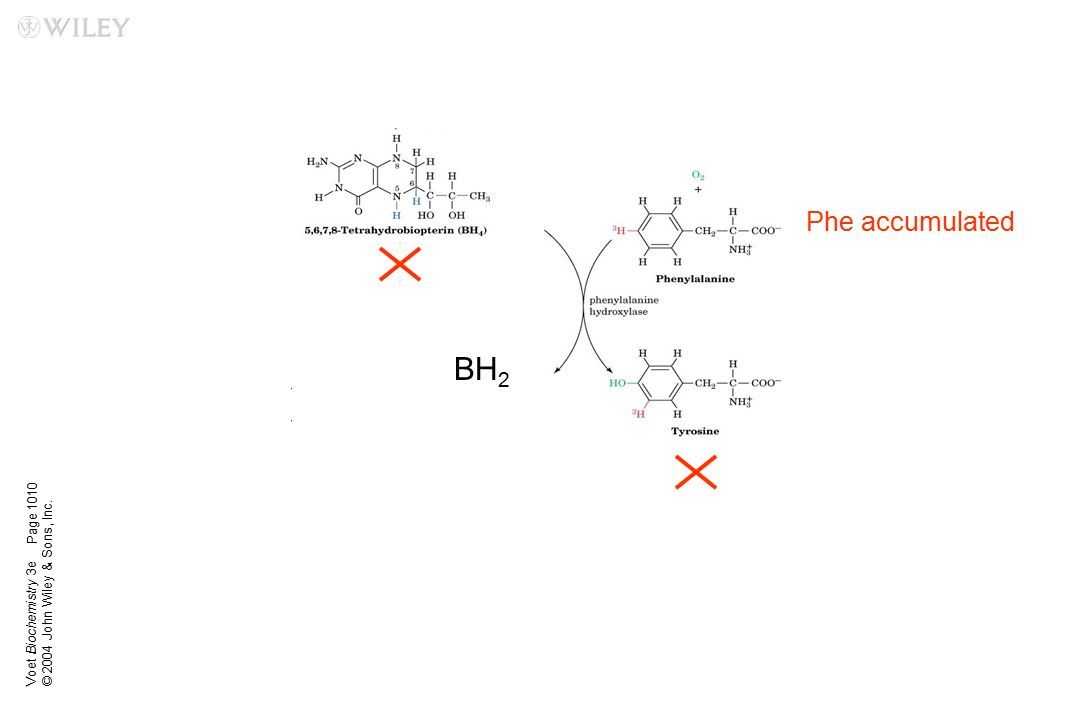 Однако исследования не показали какой-либо особой пользы тиамина, поскольку люди также принимали диету с ограничением BCAA. Хотя испытание добавок тиамина кажется разумным, клинической эффективности не наблюдалось.
Однако исследования не показали какой-либо особой пользы тиамина, поскольку люди также принимали диету с ограничением BCAA. Хотя испытание добавок тиамина кажется разумным, клинической эффективности не наблюдалось.
Есть ли факторы, которые могут повлиять на результаты лабораторных исследований? В частности, принимает ли ваш пациент какие-либо лекарства — безрецептурные или травяные — которые могут повлиять на результаты лабораторных исследований?
Аминокислоты в моче не могут адекватно количественно определять повышенные уровни BCAA.
Запах сотолона также может быть обнаружен у людей, употреблявших пажитник и любисток.Другие запахи, такие как ароматизированные подгузники или чистящие средства, также могут вызывать запах «кленового сиропа», о чем часто сообщают родители.
Copyright © 2017, 2013 ООО «Поддержка принятия решений в медицине». Все права защищены.
Ни один спонсор или рекламодатель не участвовал, не одобрял и не платил за контент, предоставляемый Decision Support in Medicine LLC. Лицензионный контент является собственностью DSM и защищен авторским правом.
Дефицит кетоацилдегидрогеназы с разветвленной цепью: болезнь кленового сиропа
Chuang DT, Shih VE: Болезнь мочи кленового сиропа (кетоацидурия с разветвленной цепью) . В Метаболические и молекулярные основы наследственного заболевания , изд. 8. Под редакцией Скривер CR, Боде А.Л., Валле Д., Слай WS. Нью-Йорк: Макгроу-Хилл; 2001.
Google Scholar
Суряван А., Хавс Дж. У., Харрис Р. А., и др. : Молекулярная модель метаболизма аминокислот с разветвленной цепью человека . Am J Clin Nutr 1998, 68 : 72–81.Исчерпывающее исследование на человеческом материале показывает распределение трансаминирования и окисления BCAA по всему телу и то, как они существенно отличаются от крыс.
PubMed
CAS
Google Scholar

Мортон Д.Х., Штраус К.А., Робинсон Д.Л., и др. : Диагностика и лечение болезни кленового сиропа: исследование 36 пациентов . Педиатрия 2002, 109 : 999–1008. В этой статье описывается ведение и исходы для большой когорты новорожденных с классическим заболеванием, прошедших проспективное лечение, с последующим наблюдением в 219 пациенто-лет.Подробно представлены принципы ведения новорожденных.
PubMed
Статья
Google Scholar
Додд П.Р., Уильямс С.Х., Гундлах А.Л., и др. : Нейромедиаторные системы глутамата и гамма-аминомасляной кислоты в острой фазе болезни мочи кленовым сиропом и цитруллинемической энцефалопатии у новорожденных телят . J Neurochem 1992, 59 : 582–590.
PubMed
Статья
CAS
Google Scholar
ДиДжордж А.М., Резвани И., Гарибальди Л.Р., Шварц M: Проспективное исследование болезни кленового сиропа и мочи в течение первых четырех дней жизни . N Engl J Med 1982, 307 : 1492–1495.
PubMed
CAS
Статья
Google Scholar
Митч В.Е., Голдберг AL: Механизмы истощения мышц: роль пути убиквитин-протеасома . N Engl J Med 1996, 335 : 1897–1905.
PubMed
Статья
CAS
Google Scholar
Томпсон Г.Н., Фрэнсис Д.Е., Халлидей D: Острое заболевание, вызванное кленовым сиропом, болезнь мочи: динамика метаболизма белков и последствия для лечения . J Pediatr 1991, 119 : 35–41.
PubMed
Статья
CAS
Google Scholar
Чанг Х.Р., Бистриан B: Роль цитокинов в катаболических последствиях инфекции и травм . JPEN J Parenter Enteral Nutr 1998, 22 : 156–166.
PubMed
CAS
Статья
Google Scholar
Kamei A, Takashima S, Chan F, Becker LE: Аномальное развитие дендритов при болезни мочи кленового сиропа . Pediatr Neurol 1992, 8 : 145–147.
PubMed
Статья
CAS
Google Scholar
Duffell SJ, Harper PA, Healy PJ, Dennis JA: Врожденный гипомиелиногенез телят герефорда . Vet Rec 1988, 123 : 423–424.
PubMed
CAS
Google Scholar
Пренски А.Л., Мозер HW: Липиды мозга, протеолипиды и свободные аминокислоты в кленовом сиропе болезнь мочи . J Neurochem 1966, 13 : 863–874.
PubMed
Статья
CAS
Google Scholar
Taketomi T, Kunishita T, Hara A, Mizushima S: Аномальный белковый и липидный состав церебрального миелина у пациента с болезнью мочи кленового сиропа . Jpn J Exp Med 1983, 53 : 109–116.
PubMed
CAS
Google Scholar
Crawford MA, Bloom M, Broadhurst CL, et al. : Доказательства уникальной функции докозагексаеновой кислоты в ходе эволюции современного мозга гоминидов . Липиды 1999, 34 (доп.) : S39-S47. Ясное и проницательное обсуждение биологической важности липидного состава головного мозга, написанное с эволюционной и антропологической, а не молекулярной точки зрения.
PubMed
Статья
CAS
Google Scholar
Базан Н.Г., Скотт БЛ: Диетические жирные кислоты омега-3 и накопление докозагексаеновой кислоты в палочковидных фоторецепторных клетках сетчатки и в синапсах . Ups J Med Sci 1990, 48 (доп.) : 97–107.
CAS
Google Scholar
Итокадзу Н., Икегая Ю., Нишикава М., Мацуки N: Двунаправленное действие докозагексаеновой кислоты на нейротрансмиссию гиппокампа in vivo . Brain Res 2000, 862 : 211–216.
PubMed
Статья
CAS
Google Scholar
Davidson BC, Cantrill RC, Kurstjens NP, Patton J: Депривация полиеновых жирных кислот у молодых кошек модулирует высвобождение 3H-дофамина пресинаптическими рецепторами в хвостатых срезах . In Vivo 1988, 2 : 295–298.
PubMed
CAS
Google Scholar
Китайка К., Пушкаш Л.Г., Звара А., и др. : Роль n-3 полиненасыщенных жирных кислот в мозге: модуляция экспрессии генов мозга крысы с помощью пищевых n-3 жирных кислот . Proc Natl Acad Sci U S A 2002, 99 : 2619–2624. Используя технологию микрочипов для классификации паттернов экспрессии информационной РНК в головном мозге, Kitajka et al. демонстрируют глубокие изменения регуляции транскрипции у животных с дефицитом омега-3 жирных кислот, при этом более 60 генов, ограниченных мозгом, демонстрируют ± трехкратное изменение экспрессии.
PubMed
Статья
CAS
Google Scholar
Champoux M, Hibbeln JR, Shannon C, et al.: Добавки жирных кислот и развитие нейромоторного аппарата у новорожденных макак-резусов . Pediatr Res 2002, 51 : 273–281. У живых приматов дефицит омега-3 жирных кислот в качестве изолированной переменной изменяет поведение зрелого животного.
PubMed
Статья
CAS
Google Scholar
Young C, Gean PW, Chiou LC, Shen YZ: Докозагексаеновая кислота ингибирует синаптическую передачу и эпилептиформную активность в гиппокампе крысы . Synapse 2000, 37 : 90–94.
PubMed
Статья
CAS
Google Scholar

Nii T, Segawa H, Taketani Y, et al. : Молекулярные события, участвующие в повышающей регуляции Na + -независимого переносчика нейтральных аминокислот LAT1 человека во время активации Т-клеток . Biochem J 2001, 358 : 693–704.
PubMed
Статья
CAS
Google Scholar
Вайнер М., Коэльо Д.М., Баршак А.Г., и др. : Снижение концентраций больших нейтральных аминокислот в плазме и спинномозговой жидкости пациентов с болезнью мочи кленового сиропа во время кризов . J Наследовать Metab Dis 2000, 23 : 505–512. Нарушение метаболизма MSD более точно описывается одновременным истощением нескольких аминокислот, незаменимых и несущественных, по мере повышения концентраций лейцина и aKIC.
PubMed
Статья
CAS
Google Scholar
Chace DH, Hillman SL, Millington DS, et al. : Быстрая диагностика болезни мочи кленовым сиропом в пятнах крови новорожденных с помощью тандемной масс-спектрометрии . Clin Chem 1995, 41 : 62–68.
PubMed
CAS
Google Scholar
Naylor EW, Guthrie R: Скрининг новорожденных на болезнь мочи кленового сиропа (кетоаацидурия с разветвленной цепью) . Pediatr 1978, 61 : 262–266.
CAS
Google Scholar
Араухо П., Вассерманн Г.Ф., Таллини К., и др. : Снижение уровней больших нейтральных аминокислот в плазме и мозге крыс с гиперлейцинемией . Neurochem Int 2001, 38 : 529–537. Тщательно разработанное исследование, демонстрирующее предсказанные явления аномального транспорта аминокислот in situ, демонстрирующее, что в дополнение к заблокированному притоку усиленный отток LNAA является механизмом их истощения из-за высокого уровня лейцина.
PubMed
Статья
CAS
Google Scholar
Banos G, Daniel PM, Moorhouse SR, Pratt OE: Ингибирование проникновения некоторых аминокислот в мозг с наблюдениями за умственной отсталостью при аминоацидурии . Psychol Med 1974, 4 : 262–269.
PubMed
CAS
Google Scholar
Юдкофф М., Дайхин Ю., Ниссим И., и др.: Ингибирование продукции глутамина астроцитами альфа-кетоизокапроновой кислотой . J Neurochem 1994, 63 : 1508–1515.
PubMed
CAS
Статья
Google Scholar
Meier C, Ristic Z, Klauser S, Verrey F: Активация гетеродимерных аминокислотных обменников системы L внутриклеточными субстратами . EMBO J 2002, 21 : 580–589.
PubMed
Статья
CAS
Google Scholar
Zielke HR, Zielke CL, Baab PJ, Collins RM: Автообмен больших нейтральных аминокислот при введении микродиализом в мозг крысы: значение для болезни мочи кленового сиропа и фенилкетонурии . Neurochem Int 2002, 40 : 347–354. Важное свойство гетерообмена или «транс-стимуляции» демонстрируется in vivo.
PubMed
Статья
Google Scholar
Smith QR, Stoll J: Транспорт аминокислот через гематоэнцефалический барьер .In Введение в гематоэнцефалический барьер: методология, биология и патология . Под редакцией Пардриджа WM. Кембридж: Издательство Кембриджского университета; 1998. Четкое и краткое описание транспорта LNAA через ГЭБ, опосредованного LAT1, написанное двумя первопроходцами в этой области. Предоставляется полный список кинетических параметров (Km, Vmax и скорость притока).
Google Scholar

Zielke HR, Huang Y, Baab PJ, et al.: Влияние альфакетоизокапроата и лейцина на окисление глутамата и глутамина in vivo в головном мозге крысы . Neurochem Res 1997, 22 : 1159–1166.
PubMed
Статья
CAS
Google Scholar
Zielke HR, Huang Y, Tildon JT, et al. : Повышение уровня аминокислот в интерстициальном пространстве мозга крысы после инфузии крупных нейтральных аминокислот и кетокислот с помощью микродиализа: инфузия альфа-кетоизокапроата . Dev Neurosci 1996, 18 : 420–425.
PubMed
CAS
Google Scholar
McManus ML, Churchwell KB, Strange K: Регулирование объема клеток при здоровье и болезни . N Engl J Med 1995, 333 : 1260–1266.
PubMed
Статья
CAS
Google Scholar
Паредес А., МакМанус М., Квон Х.М., Странный К: Осморегуляция активности котранспортера Na (+) — инозита и уровни мРНК в глиальных клетках головного мозга . Am J Physiol 1992, 263 : C1282-C1288.
PubMed
CAS
Google Scholar
Hertz L, Chen Y, Spatz M: Вовлечение ненейронных клеток головного мозга в AVP-опосредованную регуляцию водного пространства на клеточном, органном уровне и уровне всего тела . J Neurosci Res 2000, 62 : 480–490.
PubMed
Статья
CAS
Google Scholar
Franchi-Gazzola R, Visigalli R, Dall’Asta V, et al. : Истощение аминокислот активирует TonEBP и транспорт инозита, связанный с натрием . Am J Physiol Cell Physiol 2001, 280 : C1465-C1474.
PubMed
CAS
Google Scholar
Буссолати О. , Далл’Аста В., Франчи-Газзола Р., и др. : Роль системы А в транспорте нейтральных аминокислот в регуляции объема клетки . Mol Membr Biol 2001, 18 : 27–38.
, Далл’Аста В., Франчи-Газзола Р., и др. : Роль системы А в транспорте нейтральных аминокислот в регуляции объема клетки . Mol Membr Biol 2001, 18 : 27–38.
PubMed
Статья
CAS
Google Scholar
Сарфараз Д., Фрейзер CL: Влияние аргинина вазопрессина на регуляцию объема клеток в астроцитах головного мозга в культуре . Am J Physiol 1999, 276 : E596.
PubMed
CAS
Google Scholar
Guyton AC, Hall JE: Интеграция почечных механизмов для контроля объема крови и объема внеклеточной жидкости .В Учебник медицинской физиологии , изд. 9. Под редакцией Guyton AC, Hall JE. Филадельфия: WB Saunders Company; 1996.
Google Scholar
DePasquale M, Patlak CS, Cserr HF: Регулирование ионов и объема мозга во время острой гипернатриемии у крыс Brattleboro . Am J Physiol (Лондон) 1989, 256 : F1059-F1064.
CAS
Google Scholar
Кролл М., Юлер М., Линдхольм Дж .: Гипонатриемия при остром заболевании головного мозга . J Intern Med 1992, 232 : 291–297.
PubMed
CAS
Статья
Google Scholar
Кауфман S: Фенилкетонурия и ее варианты . В Тетрагидробиоптерин: основы биохимии и роль в болезнях человека . Балтимор: издательство Университета Джона Хопкинса; 1997.
Google Scholar
Пратт OE: Новый подход к лечению фенилкетонурии . J Ment Defic Res 1980, 24 : 203–217.
PubMed
CAS
Google Scholar
Surtees R, Blau N: Нейрохимия фенилкетонурии . Eur J Pediatr 2000, 159 (доп.) : S109-S113. Современный взгляд на более ранние обзоры Пратта [42] и Кауфмана [41].
Eur J Pediatr 2000, 159 (доп.) : S109-S113. Современный взгляд на более ранние обзоры Пратта [42] и Кауфмана [41].
PubMed
Статья
CAS
Google Scholar
Andrade JP, Castanheira-Vale AJ, Paz-Dias PG, et al. : Дендритные деревья нейронов из гиппокампа взрослых крыс, лишенных белка: количественное исследование Гольджи . Exp Brain Res 1996, 109 : 419–433.
PubMed
Статья
CAS
Google Scholar
Андраде Дж. П., Кастанхейра-Вале А.Дж., Мадейра, доктор медицины: Временной масштаб и степень потери нейронов и синапсов в гиппокампе у истощенных взрослых крыс . Brain Res 1996, 718 : 1–12.
PubMed
Статья
CAS
Google Scholar
Huttenlocher PR: Невропатология фенилкетонурии: исследования на людях и животных . Eur J Padiatr 2000, 159 (доп.) : S102-S106.
Артикул
Google Scholar
Ройланд Дж., Конат Г.В., Кано М., Виггинс RC: Понижающая регуляция миелин-специфичных мРНК в механизме гипомиелинизации в развивающемся головном мозге с недостаточным питанием . Brain Res Dev Brain Res 1992, 65 : 223–226.
PubMed
Статья
CAS
Google Scholar
Teicher MH, Andersen SL, Navalta CP, et al. : Психоневрологические расстройства в детском и подростковом возрасте . In Neuropsychiatry and Clinical Neurosciences , edn 4. Под редакцией Yudofsky SC, Hales RE. Вашингтон, округ Колумбия: Американское психиатрическое издательство; 2002.
Google Scholar
Lykkelund C, Nielsen JB, Lou HC, et al. : Повышенный биосинтез нейромедиаторов при фенилкетонурии, вызванный ограничением фенилаланина или дополнением неограниченной диеты большими количествами тирозина . Eur J Pediatr 1988, 148 : 238–245.
Eur J Pediatr 1988, 148 : 238–245.
PubMed
Статья
CAS
Google Scholar
Руководство по питанию при болезни кленового сиропа: подход, основанный на фактических данных и консенсусе
Основные моменты
- •
Управление питанием при MSUD было изучено на основе анализа фактических данных и консенсуса.
- •
Темы включали уровни BCAA, использование тиамина, беременность, болезни и трансплантацию.
- •
Дается объяснение онлайн-разработки руководства.
- •
Руководство включает сводные заявления, рекомендации с рейтингами и ссылками.
- •
Информация о MSUD и ресурсы для врачей и пациентов включены.
Abstract
Стремясь повысить уровень гармонизации медицинской помощи и сделать возможным изучение результатов, Международная ассоциация специалистов по генетической метаболической диете (GMDI) и Юго-восточное региональное объединение по скринингу новорожденных и генетике (SERC) сотрудничают с целью разработки руководящих принципов управления питанием для наследственных метаболических заболеваний. расстройств (IMD) с использованием модели, сочетающей методологию, основанную на доказательствах и консенсусе.Первое, что нужно выполнить, касается болезни мочи кленового сиропа (MSUD). В этом отчете описывается методология, использованная при его разработке: формулировка пяти вопросов исследования; обзор, критическая оценка и абстракция рецензируемых исследований и неопубликованной практической литературы; и вклад экспертов через опросы Delphi и номинальный групповой процесс. Этот отчет включает в себя сводные утверждения по каждому вопросу исследования и выработанные ими рекомендации по управлению питанием. За каждой рекомендацией следует стандартизированная оценка, основанная на силе используемых доказательств и консенсусе.Применение технологий для создания инфраструктуры для этого проекта обеспечило прозрачность при разработке этого руководства и станет основой для будущих руководств. Открытый онлайн-доступ к полному опубликованному руководству позволяет его использовать поставщикам медицинских услуг, исследователям и сотрудникам, которые консультируют, защищают и заботятся о лицах с MSUD и их семьях. Будут обновлены в будущем, если это будет подтверждено развитием исследований и клинической практики.
Открытый онлайн-доступ к полному опубликованному руководству позволяет его использовать поставщикам медицинских услуг, исследователям и сотрудникам, которые консультируют, защищают и заботятся о лицах с MSUD и их семьях. Будут обновлены в будущем, если это будет подтверждено развитием исследований и клинической практики.
Сокращения
BCAA
аминокислоты с разветвленной цепью
BCKA
α-кетокислоты с разветвленной цепью
BCKD
дегидрогеназа α-кетокислоты с разветвленной цепью
DRI
диетическая справочная норма
GMDI
HRSA International
Health Resources администрирование
IMD
унаследованные метаболические нарушения
MeSH
медицинская предметная рубрика
MS / MS
тандемная масс-спектрометрия
MSUD
болезнь мочи кленового сиропа
MySQL
язык структурированных запросов
NAD
никотинамид аденин динуклеотид
Национальный центр информации NCBI
NCBI
Популяция PICO
, вмешательство, сравнение и результаты
SERC
Юго-восточный региональный центр по скринингу новорожденных и генетика, совместное управление ресурсами здравоохранения и службами (HRSA), регион 3
TPP
тиаминпирофосфат
Ключевые слова
Болезнь мочи кленового сиропа
Питание изнасилование / диетотерапия
Практическое руководство
Врожденные аминокислотные патологии
Врожденные нарушения обмена веществ
Унаследованные метаболические нарушения
Рекомендуемые статьиЦитирующие статьи (0)
Просмотреть аннотацию
Copyright © 2014 Авторы.Опубликовано Elsevier Inc.
Рекомендуемые статьи
Цитирующие статьи
PRIME PubMed | Журнальные статьи о лейцинозе из PubMed
Обсуждаются результаты и значение программ массового обследования новорожденных на врожденные нарушения метаболизма, проводимых Национальным исследовательским институтом матери и ребенка (NRIMC). В 1964 году впервые в Польше был введен массовый скрининг на фенилкетонурию (ФКУ).
 В будущем мы планируем подготовить рекомендации по принципам диагностики и лечения галактоземии у детей и женщин репродуктивного возраста. Масс-скрининг на муковисцидоз также все еще обсуждается. Результаты ранних программ скрининга не были удовлетворительными, и испытания были прекращены.В 1998 г., после реорганизации всей системы, скрининг МВ с использованием трипсин-радиоиммунных тестов был снова начат. Новая программа скрининга сочетается с молекулярно-генетическим исследованием различных мутаций. Еще слишком рано оценивать важность и успех этой программы массового обследования на МВ. Мы решили прекратить скрининг на гомоцистинурию, гистидинемию, тирозинемию, лейциноз и нейробластому, поскольку эти программы не соответствовали критериям массового скрининга.В 1997 году в NRIMC была проведена серьезная реорганизация программ скрининга на врожденные нарушения метаболизма. Тест Гатри на PKU был изменен на количественный колориметрический метод. Для определения ТТГ используется иммуно-люминометрический метод. Вся система основана на полном компьютерном управлении всеми этапами скрининга, от забора крови на фильтровальной бумаге до окончательной диагностики. Обсуждаются преимущества этой современной системы организации программы скрининга.
В будущем мы планируем подготовить рекомендации по принципам диагностики и лечения галактоземии у детей и женщин репродуктивного возраста. Масс-скрининг на муковисцидоз также все еще обсуждается. Результаты ранних программ скрининга не были удовлетворительными, и испытания были прекращены.В 1998 г., после реорганизации всей системы, скрининг МВ с использованием трипсин-радиоиммунных тестов был снова начат. Новая программа скрининга сочетается с молекулярно-генетическим исследованием различных мутаций. Еще слишком рано оценивать важность и успех этой программы массового обследования на МВ. Мы решили прекратить скрининг на гомоцистинурию, гистидинемию, тирозинемию, лейциноз и нейробластому, поскольку эти программы не соответствовали критериям массового скрининга.В 1997 году в NRIMC была проведена серьезная реорганизация программ скрининга на врожденные нарушения метаболизма. Тест Гатри на PKU был изменен на количественный колориметрический метод. Для определения ТТГ используется иммуно-люминометрический метод. Вся система основана на полном компьютерном управлении всеми этапами скрининга, от забора крови на фильтровальной бумаге до окончательной диагностики. Обсуждаются преимущества этой современной системы организации программы скрининга.
6 Заболевание мочи кленовым сиропом
CASE 6
Клиническая картина
Новорожденный поступает с острой тяжелой энцефалопатией и запахом кленового сиропа (или жженого сахара).
Рисунок 6A
Рисунок 6B
Рентгенологические данные
КТ задней черепной ямки показывает низкое ослабление белого вещества мозжечка, плечевого и плечевого мостовидных суставов и покрышки связки, а также связочного отдела переднего моста с сглаживанием четвертого желудочка ( Рис. 6A1 ). Средний мозг и полушария головного мозга кажутся опухшими также с диффузно низким затуханием ( рис.6А2 ). Диффузионно-взвешенная МРТ (ДВИ) задней черепной ямки показывает заметное усиление сигнала белого вещества мозжечка, плечевого пояса, поясничной покрышки и кортикоспинальных трактов ( Рис. 6B1 ). Средний мозг опух, с повышенным сигналом DWI верхних ножек мозжечка, центрального среднего мозга, включая красные ядра, и боковой части ножек ножек, а также зрительных трактов ( Рис. 6B2 ). Вышеуказанный уровень иллюстрирует поражение боковых коленчатых тел ( рис.6B3 ), в то время как разрез, проходящий через базальные ганглии, демонстрирует двустороннее поражение задних конечностей внутренних капсул, а также прилегающих глобусов и, в некоторой степени, передне-боковых ядер таламуса ( Рис. 6B4). ).
6B1 ). Средний мозг опух, с повышенным сигналом DWI верхних ножек мозжечка, центрального среднего мозга, включая красные ядра, и боковой части ножек ножек, а также зрительных трактов ( Рис. 6B2 ). Вышеуказанный уровень иллюстрирует поражение боковых коленчатых тел ( рис.6B3 ), в то время как разрез, проходящий через базальные ганглии, демонстрирует двустороннее поражение задних конечностей внутренних капсул, а также прилегающих глобусов и, в некоторой степени, передне-боковых ядер таламуса ( Рис. 6B4). ).
Диагноз
Заболевание мочи кленовым сиропом (MSUD; также называется лейцинозом)
Дифференциальный диагноз
- Гипоксико-ишемическая энцефалопатия (ГИЭ): мозжечок и ствол мозга обычно не опухшие, характерные признаки в анамнезе
- Сепсис: отсутствие типичного паттерна MSUD
- Другое метаболическое заболевание новорожденного: отсутствие типичного паттерна MSUD
Обсуждение
Предпосылки
MSUD, также называемый лейцинозом, является заболеванием, о котором впервые сообщили в 1954 г. в семье, которая потеряла четырех младенцев в течение первых 3 месяцев жизни из-за нейродегенеративного расстройства.Моча этих младенцев имела запах, напоминающий запах кленового сиропа.
MSUD — это аминоацидопатия с разветвленной цепью, вызванная пониженной активностью дегидрогеназы α-кетокислоты с разветвленной цепью (BCKA), второго фермента на пути разложения лейцина, изолейцина и валина. Заболевание в основном связано с повышенной концентрацией лейцина, так как изолейцин и валин обладают небольшой токсичностью. Этот дефект редко встречается среди населения в целом (1/850 000), но может быть обычным явлением в некоторых закрытых сообществах, таких как поселения меннонитов (до 1/170).
Есть две формы болезни. Оба могут быть выражены в одной семье. Классическая форма характеризуется острым неонатальным предлежанием, низкой толерантностью к лейцину и очень низкой ферментативной активностью (<2,5% от контроля). Фенотип варианта менее тяжелый, с поздним началом, разумной толерантностью к лейцину и хорошей ферментативной активностью (от 7 до 20% от контроля), но вариантные формы также могут развить острое ухудшение.
Фенотип варианта менее тяжелый, с поздним началом, разумной толерантностью к лейцину и хорошей ферментативной активностью (от 7 до 20% от контроля), но вариантные формы также могут развить острое ухудшение.
Этиология
MSUD — аутосомно-рецессивное заболевание.В настоящее время известно более 50 различных мутаций в генах, которые управляют компонентами ферментов аминокислот с разветвленной цепью (BCAA) и дегидрогеназного комплекса BCKA.
Патофизиология
Болезнь мочи кленовым сиропом (MSUD)
Обзор
Что такое болезнь мочи кленового сиропа (MSUD)?
Болезнь мочи кленовым сиропом (MSUD) — это опасное для жизни нарушение обмена веществ.Нарушения обмена веществ — это состояния, при которых ваше тело не может нормально функционировать, потому что оно не может должным образом преобразовывать пищу в энергию для поддержания здоровья вашего тела.
Белок необходим организму для нормального функционирования. Белки состоят из 20 различных типов аминокислот. Белки должны расщепляться (метаболизироваться), чтобы они могли усваиваться и использоваться организмом. У людей с MSUD нет необходимых ферментов (либо у них нет определенных ферментов, либо у них есть определенные ферменты, но они не работают, либо у них недостаточно определенного фермента), чтобы расщепить три конкретных аминокислоты. кислоты — лейцин, изолейцин и валин.
Поскольку люди с MSUD не могут расщеплять эти три аминокислоты, эти аминокислоты накапливаются в организме, становятся токсичными для организма и вызывают серьезные проблемы со здоровьем. Без медицинского лечения болезнь мочи кленовым сиропом может привести к широкому спектру умственных и физических недостатков и смерти.
Какие бывают типы болезни мочи кленовым сиропом (MSUD)?
Четыре основных типа MSUD:
- Classic: Классическая болезнь мочи с кленовым сиропом является наиболее тяжелым типом MSUD.
 Тоже самое распространенное. Симптомы обычно развиваются в течение первых трех дней после рождения.
Тоже самое распространенное. Симптомы обычно развиваются в течение первых трех дней после рождения. - Промежуточный: Этот тип MSUD менее серьезен, чем классический MSUD. Симптомы обычно появляются у детей в возрасте от 5 месяцев до 7 лет.
- Прерывистый: Дети с прерывистым МСУД развиваются, как ожидалось, до тех пор, пока инфекция или период стресса не вызовут появление симптомов. Люди с прерывистым MSUD обычно могут переносить более высокие уровни трех аминокислот, чем люди с классическим MSUD.
- Тиамин-чувствительный: Этот тип MSUD реагирует на лечение с использованием высоких доз витамина B1 (тиамина) наряду с ограниченным питанием. При лечении люди с чувствительным к тиамину MSUD имеют более высокую толерантность к трем аминокислотам.
Насколько распространена болезнь мочи кленовым сиропом (MSUD)?
MSUD встречается очень редко. Это происходит примерно у 1 из каждых 185 000 рождений во всем мире. Чаще встречается в популяциях с небольшим генофондом или когда двоюродные братья и другие близкие родственники имеют вместе детей.Около 2000 человек в США живут с MSUD. Он одинаково влияет на мужчин и женщин.
Кто страдает болезнью мочи кленового сиропа (MSUD)?
MSUD может поразить любого, но люди, чьи родители находятся в близком родстве, гораздо чаще страдают метаболическим заболеванием. По этой причине MSUD часто встречается среди меннонитов в Соединенных Штатах, где члены общины часто женятся друг на друге. MSUD встречается у 1 из каждых 380 рождений в популяции меннонитов.
Симптомы и причины
Как люди заражаются болезнью мочи кленового сиропа (MSUD)?
MSUD наследуется (передается) через семьи.Ребенок рождается с MSUD, когда оба родителя являются носителями трех конкретных генных мутаций (изменений), и их ребенок наследует копии этих измененных генов — по одной копии от каждого родителя. Эти мутации практически не приводят к активности ферментов, необходимых для расщепления трех определенных аминокислот, содержащихся в продуктах, богатых белком. Эти три специфические аминокислоты — лейцин, изолейцин и валин. Без необходимых ферментов три аминокислоты накапливаются, а вместе с ними и их токсичные побочные продукты (так называемые кетокислоты).Это приводит к серьезным проблемам со здоровьем, наблюдаемым в MSUD.
Эти мутации практически не приводят к активности ферментов, необходимых для расщепления трех определенных аминокислот, содержащихся в продуктах, богатых белком. Эти три специфические аминокислоты — лейцин, изолейцин и валин. Без необходимых ферментов три аминокислоты накапливаются, а вместе с ними и их токсичные побочные продукты (так называемые кетокислоты).Это приводит к серьезным проблемам со здоровьем, наблюдаемым в MSUD.
MSUD чаще встречается в сообществах с небольшими генетическими вариациями (например, в сообществе меннонитов в США). В этих группах выше концентрация людей, которые являются носителями мутировавшего гена.
Каковы симптомы болезни мочи кленовым сиропом (MSUD)?
Симптомы классического МСУД появляются у новорожденных в течение 48 часов после рождения. У детей старшего возраста признаки промежуточного, прерывистого и чувствительного к тиамину MSUD обычно развиваются в возрасте до семи лет.Все четыре типа MSUD имеют симптомы, в том числе:
- Моча, пот или ушная сера с запахом кленового сиропа или жженым сахаром. (Это расстройство получило свое название от этого распространенного симптома.) Это не всегда может присутствовать во всех типах.
- Плохое питание, рвота, потеря аппетита, раздражительность.
- Вялость / медлительность / усталость и слабость.
- Изменения мышечного тонуса — плохой мышечный тонус, мышечная стянутость / напряжение
- Аномальные движения мышц, спазмы, вызывающие выгибание головы, шеи и позвоночника назад.
- Задержка развития.
- Припадки, судороги, дыхательная недостаточность и кома (по мере прогрессирования состояния).
Диагностика и тесты
Как диагностируется болезнь мочи кленовым сиропом (MSUD)?
Врачи диагностируют классический MSUD с помощью обследований новорожденных (анализов крови) вскоре после рождения ребенка. Люди с промежуточным, интермиттирующим или чувствительным к тиамину MSUD могут не проявлять признаков заболевания до самого раннего возраста или в раннем детстве. В этих случаях врачи ставят диагноз MSUD с помощью анализов крови и оценки симптомов ребенка, включая обнаружение характерного сахарного / кленового запаха его пота и мочи. Генетическое тестирование лейкоцитов в крови теперь может помочь подтвердить диагноз, а также определить типы MSUD.
Люди с промежуточным, интермиттирующим или чувствительным к тиамину MSUD могут не проявлять признаков заболевания до самого раннего возраста или в раннем детстве. В этих случаях врачи ставят диагноз MSUD с помощью анализов крови и оценки симптомов ребенка, включая обнаружение характерного сахарного / кленового запаха его пота и мочи. Генетическое тестирование лейкоцитов в крови теперь может помочь подтвердить диагноз, а также определить типы MSUD.
Как узнать, болен ли мой ребенок болезнью мочи кленового сиропа (MSUD)?
Если у вашего ребенка моча или пот со сладким запахом, вам следует позвонить в службу 911 или сразу обратиться в отделение неотложной помощи.Другие признаки включают слабость, вялость / усталость или внезапное снижение аппетита. Только врач может диагностировать заболевание мочи кленовым сиропом.
Ведение и лечение
Каковы методы лечения болезни мочи кленовым сиропом (MSUD)?
Врачи могут управлять MSUD, контролируя уровень трех аминокислот (лейцин, изолейцин и валин) в организме пациента.Люди с MSUD должны всегда соблюдать строгую диету, которая обеспечивает необходимые питательные вещества, но строго ограничивает количество трех аминокислот (людям действительно нужно небольшое количество из трех аминокислот для нормального роста и развития). MSUD, чувствительный к тиамину, можно контролировать с помощью высоких доз витамина B1 (тиамин) и строгой диеты.
Врачи наблюдают за людьми с MSUD на протяжении всей их жизни, чтобы убедиться, что три аминокислоты не превышают допустимый для человека уровень и не начинают причинять вред.Когда пациенты с MSUD заболевают, у них повышается температура, они не могут принимать пищу из-за рвоты или диареи или когда уровень аминокислот повышается до опасного уровня, пациента необходимо немедленно госпитализировать. В больнице врачи могут:
В больнице врачи могут:
- Введите глюкозу и инсулин через вену (через капельницу), чтобы отрегулировать уровень аминокислот в организме.
- Используйте внутривенный или назогастральный зонд для кормления (через нос) для доставки определенных питательных веществ, включая типы аминокислот, которые человек может переносить.
- Отфильтруйте плазму крови человека и верните ее в организм (процедура, называемая гемофильтрацией / диализом), чтобы снизить уровень трех аминокислот.
- Следите за пациентом на предмет признаков отека мозга, инфекции и накопления кислоты.
Существует ли постоянное лечение болезни мочи кленовым сиропом (MSUD)?
С 2004 года трансплантация печени оказалась очень успешной при лечении пациентов с классическим MSUD. С новой печенью люди с MSUD могут производить ферменты, необходимые для расщепления трех аминокислот, которые накапливаются в организме.После трансплантации печени люди с MSUD могут придерживаться неограниченной диеты, жить без симптомов и избегать дальнейших когнитивных проблем. Люди с MSUD все еще несут ген этого заболевания и, следовательно, могут передать его своим потомкам. Рекомендуется генетическое консультирование.
Каковы побочные эффекты лечения болезни мочи кленовым сиропом (MSUD)?
Побочные эффекты глюкозы и инсулина внутривенно включают изменения уровня сахара в крови. Врачи будут внимательно следить за этими уровнями в больнице, чтобы убедиться, что они остаются в пределах нормы.
Побочные эффекты гемодиализа и гемофильтрации включают мышечные судороги, низкое кровяное давление (гипотензию) и изменения содержания электролитов в крови, которые требуют тщательного наблюдения.
Пересадка печени включает осложнения, аналогичные любой хирургической процедуре, включая кровотечение, инфекцию и образование тромбов. Трансплантация также приносит другие потенциальные осложнения, включая отторжение органа и ослабление иммунной системы (помимо того, что необходимо для предотвращения отторжения органа). С улучшенным пониманием процесса трансплантации пациенты могут вести нормальную жизнь при хорошем наблюдении и не опасаясь проблем, связанных с MSUD.
Какие осложнения связаны с болезнью мочи кленовым сиропом (MSUD)?
У людей с MSUD могут развиться различные осложнения, от легких до тяжелых. Осложнения болезни мочи кленовым сиропом включают:
- Повреждение головного мозга, неврологические проблемы и задержка развития.
- Повышенный риск синдрома дефицита внимания / гиперактивности (СДВГ), беспокойства и депрессии.
- Потеря костной массы, из-за которой кости легко ломаются.
- Набухание поджелудочной железы / панкреатит, особенно во время критических состояний.
- Хронические головные боли, вызванные повышением артериального давления в черепе.
- Двигательные расстройства, такие как тремор и неконтролируемые мышечные сокращения.
- Кома и смерть в результате кризисных состояний, таких как инфекция, стресс или плохой контроль питания.
Профилактика
Как можно предотвратить болезнь мочи кленовым сиропом (MSUD)?
Невозможно предотвратить болезнь мочи кленовым сиропом.Вы можете снизить риск рождения ребенка с MSUD, убедившись, что ваш партнер не связан с вами. Если у вас есть братья и сестры или другие родственники с MSUD, вам следует поговорить со своим врачом, прежде чем забеременеть, чтобы обсудить возможность передачи болезни вашему ребенку. Ваш врач может проверить вас и вашего партнера, чтобы определить, являетесь ли вы носителем гена, вызывающего MSUD.
Что я могу сделать, чтобы облегчить симптомы болезни мочи кленового сиропа (MSUD)?
Если у вашего ребенка есть симптомы MSUD, вам не следует пытаться ухаживать за ними дома.Позвоните по номеру 911 или сразу в отделение неотложной помощи.
Перспективы / Прогноз
Каковы перспективы для пациентов с болезнью мочи кленового сиропа (MSUD)?
Люди с MSUD могут стать взрослыми, соблюдая строгую диету и избегая болезней и стрессов, насколько это возможно.Хотя с этим расстройством можно справиться с помощью диеты с ограничением белка и под тщательным медицинским наблюдением, люди с MSUD всегда подвержены риску метаболического приступа. Симптомы могут внезапно вернуться в любой момент, обычно после человека:
- Заражает.
- Не ест длительное время (постится).
- Переживает травму или стресс (физический или психологический).
- Изменил диету или не соблюдает рекомендованную диету.
При метаболическом приступе люди с MSUD должны немедленно обратиться за медицинской помощью.Немедленная помощь может снизить риск повреждения головного мозга и других осложнений. Поговорите со своим врачом о возможности трансплантации печени.
Жить с
Когда мне следует позвонить своему врачу по поводу болезни мочи кленового сиропа (MSUD)?
Если у вашего ребенка проявляются признаки MSUD, вам следует немедленно обратиться за медицинской помощью.Хотя у детей старшего возраста и взрослых заболевание развивается очень редко, вам следует обращаться к врачу каждый раз, когда вы обнаруживаете запах кленового сиропа в моче или поте.
.