
Артрогрипоз дистальный: Дистальный артрогрипоз у новорожденного: клинический случай

Артрогрипоз дистальный (синдром Фримена-Шелдона), MYh4 ч.м.
Метод определения
Секвенирование
Исследуемый материал
Цельная кровь (с ЭДТА)
Доступен выезд на дом
Исследование частых мутаций в гене MYh4.
Тип наследования.
Аутосомно- доминантный.
Гены, ответственные за развитие заболевания.
Ген MYh4 (MYOSIN, HEAVY CHAIN 3, SKELETAL MUSCLE, EMBRYONIC) расположен на хромосоме 17 в регионе 17p13.1. Мутации в данном гене приводят также к развитию артрогрипоза дистального тип 2B.
Определение заболевания.
Синдром Фримена-Шелдона –заболевание, характеризующееся наличием двух и более врожденных контрактур (стойких ограничений движений в суставе) дистальных частей конечностей.
Патогенез и клиническая картина.
Ген MYh4 кодирует тяжелую цепь 3 миозина скелетных мышц эмбриона. Синдром Фримена-Шелдона — это заболевание, касающееся патологии мышц, которое не относится к общепринятым миопатиям, но его причиной является мутации в генах, кодирующих белки сократительного аппарата медленно сокращающихся миофибрилл. Пораженные дети часто имеют дополнительную деформацию конечностей (ульнарную девиацию кистей) и характерную контрактуру мимических мышц. Такая контрактура возникает в результате очень малого размера ротового отверстия — это и послужило основой для исторического названия синдрома- «синдром свистящего лица». Дополнительными признаками лицевого фенотипа являются антимонголоидный разрез глаз, выступающие носогубные складки, гипоплазия крыльев носа, длинный фильтр, «собранные в трубочку» губы, маленький рот и Н-образная складочка (ямочка) на подбородке. У больных также отмечают глубоко посаженные глаза с гипертелоризмом (увеличенное расстояние между внутренними краями глазниц), увеличенную длину фильтра (вертикальная бороздка в средней части верхней губы от нижненосовой точки до красной каймы), маленький нос и ноздри. Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С. И., Демикова Н. С.
 Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с. - Antley, R. M., Uga, N., Burzynski, N. J., Baum, R. S., Bixler, D. Diagnostic criteria for the whistling face syndrome. Birth Defects Orig. Art. Ser. XI(5): 161-168, 1975.
- OMIM.
Артрогрипоз дистальный (синдром Фримена-Шелдона), MYh4 ч.м.
Метод определения
Секвенирование
Исследование мутаций в гене MYh4.
Тип наследования.
Аутосомно- доминантный.
Гены, ответственные за развитие заболевания.
Ген MYh4 (MYOSIN, HEAVY CHAIN 3, SKELETAL MUSCLE, EMBRYONIC) расположен на хромосоме 17 в регионе 17p13.1. Мутации в данном гене приводят также к развитию артрогрипоза дистального тип 2B.
Определение заболевания.
Синдром Фримена-Шелдона –заболевание, характеризующееся наличием двух и более врожденных контрактур (стойких ограничений движений в суставе) дистальных частей конечностей.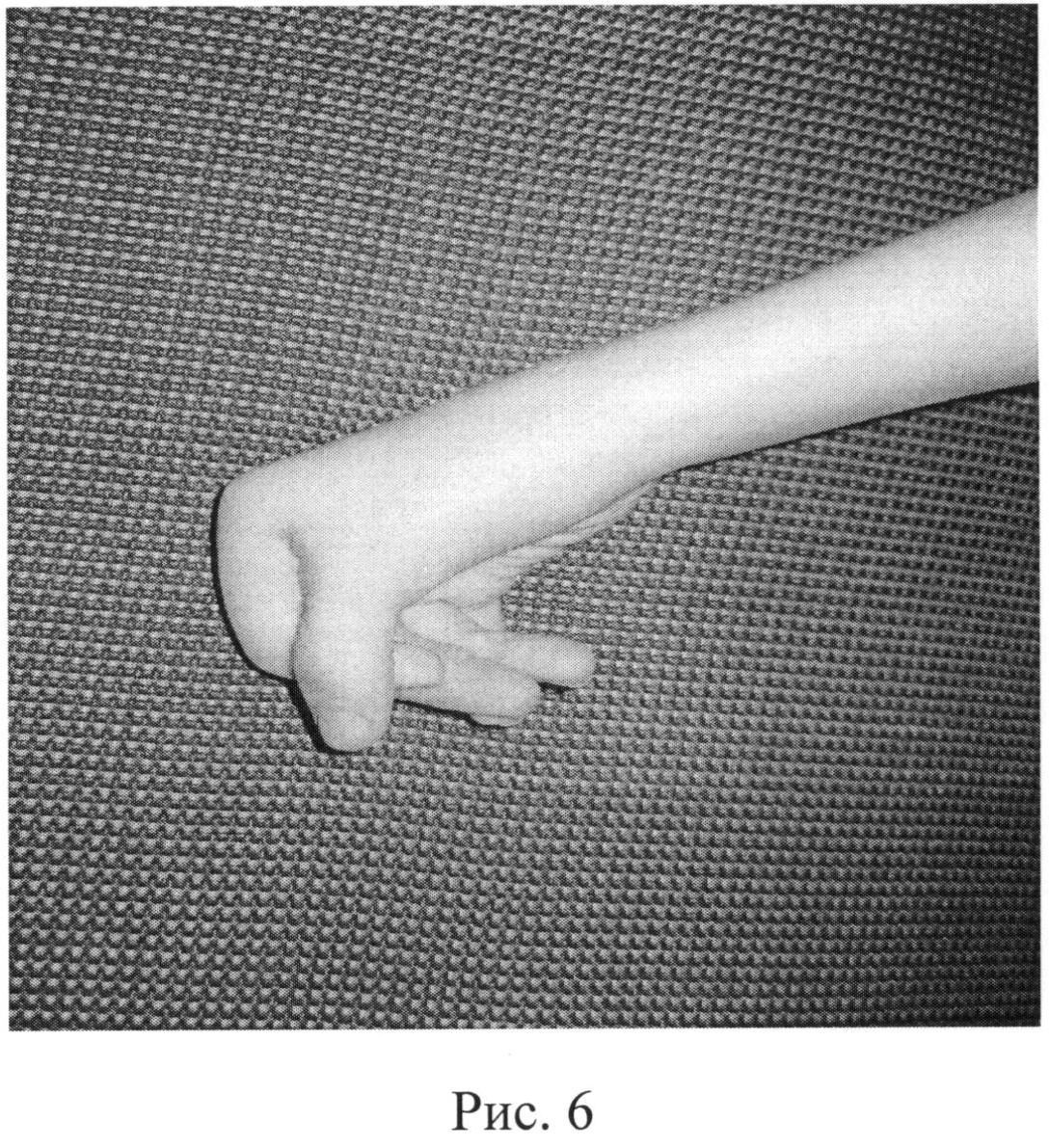
Патогенез и клиническая картина.
Ген MYh4 кодирует тяжелую цепь 3 миозина скелетных мышц эмбриона. Синдром Фримена-Шелдона — это заболевание, касающееся патологии мышц, которое не относится к общепринятым миопатиям, но его причиной является мутации в генах, кодирующих белки сократительного аппарата медленно сокращающихся миофибрилл. Пораженные дети часто имеют дополнительную деформацию конечностей (ульнарную девиацию кистей) и характерную контрактуру мимических мышц. Такая контрактура возникает в результате очень малого размера ротового отверстия — это и послужило основой для исторического названия синдрома- «синдром свистящего лица». Дополнительными признаками лицевого фенотипа являются антимонголоидный разрез глаз, выступающие носогубные складки, гипоплазия крыльев носа, длинный фильтр, «собранные в трубочку» губы, маленький рот и Н-образная складочка (ямочка) на подбородке. У больных также отмечают глубоко посаженные глаза с гипертелоризмом (увеличенное расстояние между внутренними краями глазниц), увеличенную длину фильтра (вертикальная бороздка в средней части верхней губы от нижненосовой точки до красной каймы), маленький нос и ноздри. Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С. И., Демикова Н.
 С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с. - Antley, R. M., Uga, N., Burzynski, N. J., Baum, R. S., Bixler, D. Diagnostic criteria for the whistling face syndrome. Birth Defects Orig. Art. Ser. XI(5): 161-168, 1975.
- OMIM.
Артрогрипоз дистальный (синдром Фримена-Шелдона). Поиск частых мутаций в гене MYh4, ч. м. (Arthrogryposis Distal Type 2A, Gene MYh4, Freq. Mut.)
Метод определения
Секвенирование
Исследование мутаций в гене MYh4.
Тип наследования.
Аутосомно- доминантный.
Гены, ответственные за развитие заболевания.
Ген MYh4 (MYOSIN, HEAVY CHAIN 3, SKELETAL MUSCLE, EMBRYONIC) расположен на хромосоме 17 в регионе 17p13.1. Мутации в данном гене приводят также к развитию артрогрипоза дистального тип 2B.
Определение заболевания.
Синдром Фримена-Шелдона –заболевание, характеризующееся наличием двух и более врожденных контрактур (стойких ограничений движений в суставе) дистальных частей конечностей.
Патогенез и клиническая картина.
Ген MYh4 кодирует тяжелую цепь 3 миозина скелетных мышц эмбриона. Синдром Фримена-Шелдона — это заболевание, касающееся патологии мышц, которое не относится к общепринятым миопатиям, но его причиной является мутации в генах, кодирующих белки сократительного аппарата медленно сокращающихся миофибрилл. Пораженные дети часто имеют дополнительную деформацию конечностей (ульнарную девиацию кистей) и характерную контрактуру мимических мышц. Такая контрактура возникает в результате очень малого размера ротового отверстия — это и послужило основой для исторического названия синдрома- «синдром свистящего лица». Дополнительными признаками лицевого фенотипа являются антимонголоидный разрез глаз, выступающие носогубные складки, гипоплазия крыльев носа, длинный фильтр, «собранные в трубочку» губы, маленький рот и Н-образная складочка (ямочка) на подбородке. У больных также отмечают глубоко посаженные глаза с гипертелоризмом (увеличенное расстояние между внутренними краями глазниц), увеличенную длину фильтра (вертикальная бороздка в средней части верхней губы от нижненосовой точки до красной каймы), маленький нос и ноздри. Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Задержка роста, начинающаяся после рождения, наблюдается в 62%, умственная отсталость в 31% случаев. Скелетные мальформации – камптодактилия (сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти) с девиацией локтевой кости, эквиноварусная стопа (сочетание конской стопы и внутренней косолапости), кифосколиоз, контрактура тазобедренных и коленных суставов, на рентгенограмме круто наклоненное дно передней черепной ямки. Постоянный признак синдрома — ульнарная девиация пальцев — связан не с костными нарушениями, а с поражением либо локтевого нерва, либо соответствующих мотонейронов. Пороки развития внутренних органов не характерны.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С. И., Демикова Н.
 С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с. - Antley, R. M., Uga, N., Burzynski, N. J., Baum, R. S., Bixler, D. Diagnostic criteria for the whistling face syndrome. Birth Defects Orig. Art. Ser. XI(5): 161-168, 1975.
- OMIM.
| 1 | X-сцепленная адренолейкодистрофия |
| 2 | ААА синдром, Оллгрова синдром (ахалазия, алакримия, недостаточность надпочечников |
| 3 | Аарскога-Скотта cиндром |
| 4 | Абиотрофия сетчатки, тип Франческетти |
| 5 | Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) |
| 6 | Азооспермия |
| 7 | Айкарди-Гутьереса синдром |
| 8 | Акродерматит энтеропатический |
| 9 | Аксенфельда-Ригера синдром |
| 10 | Алажиля синдром |
| 11 | Александера болезнь |
| 12 | Альбинизм глазокожный |
| 13 | Алькаптонурия |
| 14 | Альстрема синдром |
| 15 | Аменорея |
| 16 | Альфа-1-антитрипсина недостаточность |
| 17 | Ангельмана синдром |
| 18 | Андерсена синдром |
| 19 | Анемия Даймонда-Блекфена |
| 20 | Анеуплоидии |
| 21 | Аниридия |
| 22 | Антли-Бикслера синдром |
| 23 | Апера синдром |
| 24 | Арта cиндром |
| 25 | Артрогрипоз дистальный (синдром Фримена-Шелдона) |
| 26 | Атаксия Фридрейха |
| 27 | Атаксия, хорея, судороги и деменция |
| 28 | Атрофия зрительного нерва Лебера |
| 29 | Атрофия зрительного нерва с глухотой |
| 30 | Аутоиммунный лимфопролиферативный синдром |
| 31 | Аутоиммунный полиграндулярный синдром I типа |
| 32 | Аутоимунный полиэндокринный синдром |
| 33 | Афазия первичная прогрессирующая |
| 34 | Ахондроплазия |
| 35 | Баллера-Герольда синдром |
| 36 | Банаян-Райли-Рувалькаба cиндром |
| 37 | Барде-Бидля (Ларенса-Муна) синдром |
| 38 | Барта cиндром |
| 39 | Барттера синдром |
| 40 | Бёрта-Хога-Дьюба синдром |
| 41 | Бесплодие |
| 42 | Беста болезнь |
| 43 | Биотинидазы недостаточность |
| 44 | Блефарофимоз, обратный эпикант и птоз |
| 45 | Блоха-Сульцбергера синдром |
| 46 | Блума синдром |
| 47 | Боковой амиотрофический склероз |
| 48 | Боуэна-Конради синдром |
| 49 | Бранхио-окуло-фациальный синдром |
| 51 | Брахидактилия |
| 53 | Бьёрнстада синдром |
| 54 | Ваарденбурга синдром |
| 55 | Ваарденбурга-Шаха синдром |
| 56 | Ван дер Вуда синдром |
| 57 | Велокардиофациальный синдром |
| 58 | Вернера синдром |
| 59 | Видеманна-Беквита синдром, спорадическая нефробластома |
| 60 | Виллебранда болезнь |
| 61 | Вильсона-Коновалова болезнь |
| 62 | Вильямса cиндром |
| 63 | Вискотта-Олдрича cиндром |
| 64 | Вольмана болезнь, болезнь накопления эфиров холестерина |
| 65 | Вольфа-Хиршхорна синдром |
| 67 | Врожденная нечувствительность к боли с ангидрозом (врожденная сенсорная нейропатия с ангидрозом, HSAN4, CIPA) |
| 68 | Врожденной центральной гиповентиляции синдром |
| 69 | Вульгарный ихтиоз |
| 70 | Галактоземия тип I |
| 71 | Галактоземия тип II |
| 72 | Галактоземия тип III |
| 73 | Галактосиалидоз |
| 74 | Галлервордена-Шпатца болезнь |
| 75 | Ганглиозидоз GM1 тип 1,2,3 |
| 76 | Гастроинтестинальный полипоз |
| 77 | Гелеофизическая дисплазия |
| 78 | Гемофилия |
| 79 | Гемохроматоз наследственный |
| 80 | Генитопателлярный синдром |
| 81 | Германски-Пудлака синдром |
| 82 | Герстманна-Штреусслера-Шейнкера болезнь |
| 83 | Гидроцефалия, обусловленная врожденнным стенозом Сильвиева водопровода |
| 84 | Гипер-IgD синдром |
| 85 | Гипер-IgM синдром |
| 86 | Гиперкалиемический периодический паралич |
| 87 | Гипероксалурия тип I |
| 88 | Гиперорнитинемии-гипераммониемии-гомоцитрулинурии синдром (ННН синдром) |
| 89 | Гипертрофическая кардиомиопатия |
| 90 | Гиперфенилаланинемия с дефицитом тетрагидробиоптерина |
| 91 | Гиперхолестеринемии |
| 92 | Гипогонадизм |
| 93 | Гипокалиемический периодический паралич |
| 94 | Гипоспадия |
| 95 | Гипотрихоз |
| 96 | Гипофосфатазия |
| 97 | Гипофосфатемический рахит |
| 98 | Гипохондроплазия |
| 99 | Гиппеля-Линдау синдром |
| 100 | Глазо-зубо-пальцевой синдром |
| 101 | Глаукома врожденная |
| 102 | Глаукома ювенильная открытоугольная |
| 103 | Гликогеноз 0 тип |
| 104 | Гликогеноз III типа |
| 105 | Гликогеноз IV типа |
| 106 | Гликогеноз IX типа |
| 107 | Гликогеноз Iа тип |
| 108 | Гликогеноз Iв тип |
| 109 | Гликогеноз V типа |
| 110 | Гликогеноз VI типа |
| 111 | Гликогеноз XI типа, Фанкони-Бикеля синдром |
| 112 | Гломеруоцитоз почек гипопластического типа |
| 113 | Глутаровая ацидурия тип 1 |
| 114 | Глутаровая ацидурия тип 2 |
| 116 | Гомоцистинурия |
| 117 | Гоше болезнь тип 1,2,3 |
| 118 | Грейга cиндром |
| 119 | Грисцелли cиндром |
| 120 | Дауна cиндром |
| 121 | Делеции хромосомы 1p36 синдром |
| 122 | Десмоидные опухоли |
| 123 | Дефицит гормона гипофиза, комбинированный |
| 124 | Дефицит иммуноглобулина A |
| 125 | Дефицит карнитина системный первичный |
| 126 | Дефицит фактора F12 |
| 127 | Джексона-Вейсса cиндром |
| 128 | Ди Джорджи cиндром |
| 129 | Диастрофическая дисплазия |
| 130 | Дисгенезия гонад |
| 131 | Дисплазия де ля Шапеля (Ателостеогенез) |
| 132 | Дисплазия Книста |
| 133 | Дистальная моторная нейропатия |
| 134 | Дистальная спинальная амиотрофия врожденная с параличом диафрагмы |
| 135 | Дисхондростеоз Лери-Вейлля |
| 136 | Дорфмана-Чанарина синдром |
| 137 | Жильбера cиндром |
| 138 | Жубер cиндром |
| 139 | Задержка полового созревания |
| 140 | Зандхоффа болезнь |
| 141 | Изовалериановая ацидемия |
| 142 | Инверсия пола |
| 143 | Ихтиоз буллезный |
| 144 | Ихтиоз врожденный аутосомно-рецессивный |
| 145 | Ихтиоз вульгарный |
| 146 | Ихтиоз, спастическая квадриплегия и умственная отсталость |
| 147 | Кампомелическая дисплазия |
| 148 | Канавана болезнь |
| 149 | Карбамолфосфатсинтетазы недостаточность |
| 150 | Карпентера cиндром |
| 151 | Кератита-ихтиоза-тугоухости cиндром |
| 152 | Кернса-Сейра синдром |
| 153 | Клайнфельтера cиндром |
| 154 | Клиппеля-Фейля cиндром |
| 155 | Коккейна cиндром |
| 156 | Комбинированный дефицит витамин K-зависимых факторов свертывания крови |
| 157 | Косолапость врожденная с или без дефицита длинных костей и/или зеркальной полидактилией |
| 158 | Костелло cиндром |
| 159 | Костная гетероплазия прогрессирующая |
| 160 | Коудена болезнь |
| 161 | Коффина-Лоури синдром |
| 162 | Кошачьего глаза синдром |
| 163 | Кошачьего крика синдром |
| 164 | Краббе болезнь |
| 165 | Краниометафизарная дисплазия |
| 166 | Краниосиностоз |
| 167 | Краниофациальной дисморфии-глухоты-ульнарной девиации кистей синдром |
| 168 | Крейтцфельда-Якоба болезнь |
| 169 | Криглера-Найара синдром |
| 170 | Крипторхизм |
| 171 | Крузона с черным акантозом синдром |
| 172 | Крузона синдром |
| 173 | Куррарино синдром |
| 174 | Ларинго-онихо-кутанный синдром |
| 175 | Лейкодистрофия с гипомиелинизацией |
| 176 | Лейкоэнцефалопатия с «исчезающим» белым веществом, детская атаксия с гипомиелинизацией |
| 177 | Лейкоэнцефалопатия с пораженим ствола мозга и высоким уровнем лактата при спектроскопии |
| 178 | Лейкоэнцефалопатия с субкортикальными кистами |
| 179 | Лейциноз (болезнь «с запахом кленового сиропа мочи» |
| 180 | Лермитт-Дуклос болезнь |
| 181 | Леша-Найяна синдром |
| 182 | Ли синдром |
| 183 | Ли-Фраумени синдром |
| 184 | Линча синдром (наследственный неполипозный рак толстой кишки) |
| 185 | Липодистрофия врожденная генерализованная |
| 186 | Липодистрофия семейная частичная |
| 187 | Липопротеин липазы недостаточность |
| 188 | Лоу синдром |
| 189 | Люджина — Фринса синдром |
| 190 | Макла-Уэллса синдром |
| 191 | Маклеода синдром |
| 192 | Малан синдром |
| 193 | Мандибулоакральная дисплазия с липодистрофией |
| 194 | Маннозидоз альфа |
| 195 | Маринеску-Шегрена синдром |
| 196 | Мартина-Белл, УО FRAXA Синдром |
| 197 | Маршалла-Смита синдром |
| 198 | Мевалоновая ацидурия |
| 200 | Метатропная дисплазия (OMIM 156530) |
| 201 | Метахроматическая лейкодистрофия |
| 202 | Метгемоглобинемия |
| 203 | Метилмалоновая ацидурия |
| 204 | Микрофтальм изолированный |
| 205 | Микрофтальм с катарактой |
| 206 | Микроцефалии с капиллярными мальформациями синдром |
| 207 | Миллера-Дикера синдром |
| 208 | Милроя болезнь (лимфедема наследственная) |
| 209 | Миоклоническая дистония |
| 210 | Миоклоническая эпилепсия Лабофа |
| 211 | Миопатия Броди |
| 212 | Миопатия Миоши |
| 213 | Миотоническая дистрофия |
| 214 | Миотония Томсена/Беккера |
| 215 | Митохондриальные гепатопатии |
| 216 | Митохондриальные заболевания, связанные с мутациями в гене POLG |
| 217 | Митохондриальные энцефаломиопатии, связанные с мутациями мтДНК |
| 218 | Митохондриальные энцефаломиопатии, связанные с мутациями ядерных генов |
| 219 | Множественная сульфатазная недостаточность |
| 220 | Множественной эндокринной неоплазии второго типа (МЭН2) cиндром |
| 221 | Множественные вывихи суставов, задержка роста, черепно-лицевые аномалии и врожденные пороки сердца |
| 222 | Множественных птеригиумов синдром |
| 223 | Множественных синостозов синдром |
| 224 | Молибденового кофактора недостаточность |
| 225 | Монилетрикс |
| 226 | Моуат-Вильсон cиндром |
| 227 | Муковисцидоз |
| 228 | Муколипидоз II, III типа |
| 229 | Мукополисахаридоз I типа |
| 230 | Мукополисахаридоз II типа |
| 231 | Мукополисахаридоз III А, В, С, D типа |
| 232 | Мукополисахаридоз IV A, B типа |
| 233 | Мукополисахаридоз VI типа |
| 234 | Мукополисахаридоз VII типа |
| 235 | Мышечная дистрофия врождённая, интегрин А7 негативная |
| 236 | Мышечная дистрофия врожденная, мерозин-негативная |
| 237 | Мышечная дистрофия врожденная, тип 1C |
| 238 | Мышечная дистрофия Дюшенна/Беккера |
| 239 | Мышечная дистрофия поясноконечностная |
| 240 | Мышечная дистрофия тип Фукуяма |
| 241 | Мышечная дистрофия Эмери-Дрейфуса |
| 242 | Мюнке синдром |
| 243 | Накопление нейтральных липидов с миопатией |
| 244 | Нарушение формирования пола |
| 245 | Нанизм MULIBRAY |
| 246 | Нарушения гликозилирования тип 1a, синдром Жакена |
| 247 | Нарушения гликозилирования тип Ib (ген MPI) |
| 248 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I |
| 249 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II |
| 250 | Наследственная нейропатия с подверженностью параличу от сдавления |
| 251 | Наследственная оптическая нейропатия Лебера |
| 252 | Наследственные глаукомы, аномалия Петерса, дермоид роговицы |
| 253 | Наследственный амилоидоз |
| 254 | Наследственный ангионевротический отек |
| 255 | Наследственный панкреатит |
| 256 | Невынашивание беременности |
| 257 | Наследственный рак желудка |
| 258 | Недостаточность N-ацетилглютаматсинтазы |
| 259 | Недостаточность длинноцепочечной 3-гидроксиацил-КоА-дегидрогеназы жирных кислот |
| 260 | Недостаточность короткоцепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 261 | Недостаточность очень длинноцепочечной ацил-КоА дегидрогеназы жирных кислот |
| 262 | Недостаточность синтетазы голокарбоксилаз |
| 263 | Недостаточность среднецепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 264 | Недостаточность сукцинил-КоА:3-кетоацил-КоА трансферазы |
| 265 | Незаращение родничков |
| 266 | Нейроаксональная дистрофия |
| 267 | Нейродегенерация с накоплением железа 4 |
| 268 | Нейромиотония и аксональная нейропатия |
| 269 | Нейрональный цероидный липофусциноз тип 1 |
| 270 | Нейрональный цероидный липофусциноз тип 2 |
| 271 | Нейросенсорная несиндромальная тугоухость |
| 272 | Нейрофиброматоз 1 и 2 типов |
| 273 | Нейтропения тяжёлая врождённая |
| 274 | Некетотическая гиперглицинемия |
| 275 | Некомпактного левого желудочка cиндром |
| 276 | Немалиновая миопатия |
| 277 | Нефронофтиз |
| 278 | Нефротический синдром |
| 279 | Ниймеген cиндром |
| 280 | Ниманна-Пика тип А и В болезнь |
| 281 | Ниманна-Пика тип С болезнь |
| 282 | Ногтей-надколенника синдром |
| 283 | Норри болезнь |
| 284 | Нунан синдром |
| 285 | Олигозооспермия тяжелой степени |
| 286 | Окулофарингеальная мышечная дистрофия |
| 287 | Опица GBBB синдром |
| 288 | Опица-Каведжиа синдром |
| 289 | Опухоль Вильмса |
| 290 | Орнитинтранскарбамилазы недостаточность |
| 291 | Ослера-Рендю-Вебера cиндром |
| 292 | Остеолиз карпотарзальный, мультицентрический |
| 293 | Остеопетроз рецессивный (мраморная болезнь костей) |
| 294 | Паллистера-Киллиана cиндром |
| 295 | Паллистера-Холла cиндром |
| 296 | Палочко-колбочковая дистрофия |
| 297 | Пантотенат киназы недостаточность |
| 298 | Парамиотония Эйленбурга |
| 299 | Патау cиндром |
| 300 | Пейтца-Егерса синдром |
| 301 | Пелицеуса-Мерцбахера болезнь |
| 302 | Пендреда Синдром |
| 303 | Первичная аутосомно-рецессивная микроцефалия, тип 5 |
| 304 | Первичная гипертрофическая остеоартропатия (пахидермопериостоз) |
| 305 | Первичная легочная гипертензия |
| 306 | Периодическая болезнь |
| 307 | Пигментная дегенерация сетчатки |
| 308 | Пикнодизостоз |
| 309 | Пирсона синдром |
| 310 | Пневмоторакс первичный спонтанный |
| 311 | Подколенного птеригиума cиндром |
| 312 | Полидактилия |
| 313 | Поликистоз почек |
| 314 | Помпе болезнь |
| 315 | Понтоцеребеллярная гипоплазия |
| 316 | Потоцки-Лупски cиндром |
| 317 | Почечная адисплазия |
| 318 | Прадера-Вилли Синдром |
| 319 | Преждевременная недостаточность яичников |
| 320 | Прогерия Хатчинсона-Гилфорда |
| 321 | Прогрессирующая наружная офтальмоплегия, АД и АР |
| 322 | Пропионовая ацидемия |
| 323 | Псевдоахондроплазия |
| 324 | Псевдоксантома эластическая |
| 325 | Пфайффера cиндром |
| 326 | Рабдомиолиз (миоглобинурия) |
| 327 | Рак молочной железы |
| 328 | Рак почки |
| 329 | Рак щитовидной железы.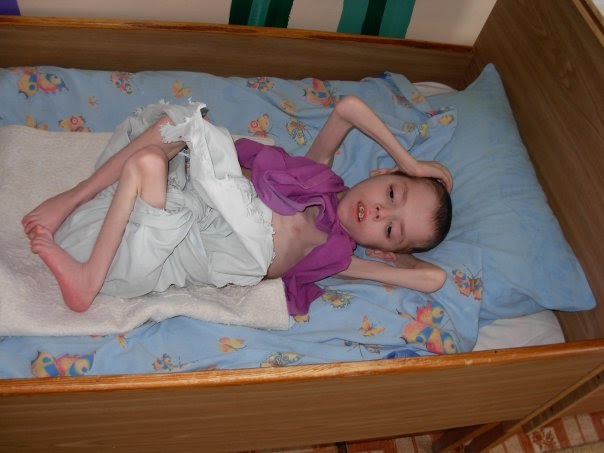 Синдром множественной эндокринной неоплазии второго типа (МЭН2) Синдром множественной эндокринной неоплазии второго типа (МЭН2) |
| 330 | Рак яичников |
| 331 | Ретинобластома |
| 332 | Ретиношизис |
| 333 | Ретта синдром |
| 334 | Рефсума болезнь |
| 335 | Ригидного позвоночника cиндром |
| 336 | Робинова синдром |
| 337 | Ротмунда-Томсена синдром |
| 338 | Рубинштейна-Тейби синдром |
| 339 | Семейная периодическая лихорадка |
| 340 | Семейный аденомоматозный полипоз, полипозный рак толстой кишки |
| 341 | Семейный внутрипеченочный холестаз 1 типа |
| 342 | Семейный внутрипеченочный холестаз 2 типа ( Баллера болезнь) |
| 343 | Семейный внутрипеченочный холестаз 3 типа |
| 344 | Семейный гемофагоцитарный лимфогистиоцитоз |
| 345 | Семейный медуллярный рак щитовидной железы |
| 346 | Семейный рак толстой кишки |
| 347 | Семейный холодовой аутовоспалительный синдром |
| 348 | Сениора-Локена синдром |
| 349 | Сенсорная полинейропатия (врожденная нечувствительность к боли) |
| 350 | Септо-оптическая дисплазия |
| 351 | Сетре-Чотзена синдром |
| 352 | Сиалидоз тип 1,2 |
| 353 | Сильвера-Рассела Синдром |
| 354 | Симпсона-Голаби-Бемель синдром |
| 355 | Синдром CADASIL, энцефалопатия с субкортикальными инфарктами |
| 357 | Синдром CINCA (холодовая лихорадка, синдром Мукле-Велса) |
| 358 | Синдром CRASH |
| 359 | Синдром ESC |
| 360 | Синдром LEOPARD |
| 361 | Синдром MASA |
| 362 | Синдром MNGIE |
| 363 | Синдром Ohdo, SBBYSS вариант |
| 364 | Синдром RAPADILINO |
| 365 | Синдром TAR |
| 366 | Синдром TRAPS (злокачественная гипертермия, амилоидоз почек) |
| 367 | Синдром тугоухости и атрофии зрительных нервов |
| 368 | Скапулоперонеальная миопатия |
| 370 | Смита-Лемли-Опитца синдром |
| 371 | Смит-Магенис синдром |
| 372 | Сотоса синдром |
| 373 | Спастическая параплегия Штрюмпеля |
| 374 | Спинальная амиотрофия типы I, II, III, IV |
| 375 | Спинальная и бульбарная амиотрофия Кеннеди |
| 376 | Спиноцеребеллярная атаксия |
| 377 | Спонгиоформная энцефалопатия с нейропсихическими проявлениями |
| 378 | Спондилокостальный дизостоз |
| 379 | Спондилоэпифизарная дисплазия (SEDT) |
| 380 | Стиклера синдром |
| 381 | Суперактивность фосфорибозилпирофосфат синтетазы |
| 382 | Талассемия beta |
| 383 | Тестикулярной феминизации синдром |
| 384 | Тея-Сакса болезнь |
| 385 | Тирозингидроксилазы недостаточность |
| 386 | Тирозинемия тип I |
| 387 | Торсионная дистония |
| 388 | Транспортера глюкозы недостаточность |
| 389 | Трихоринофалангеальный синдром |
| 390 | Тричера Коллинза-Франческетти синдром |
| 391 | Тромбоцитопения врожденная |
| 392 | Туберозный склероз |
| 393 | Умственная отсталость моногенная |
| 394 | Унферрихта-Лундборга болезнь |
| 395 | Уокера-Варбург синдром |
| 396 | Ушера синдром |
| 397 | Фабри болезнь |
| 398 | Фатальная семейная инсомния |
| 399 | Фацио-Лонде болезнь |
| 400 | Фелан-МакДермид синдром |
| 401 | Фенилкетонурия |
| 402 | Фибродисплазия оссифицирующая прогрессирующая |
| 403 | Фокальная кожная гипоплазия (Горлина-Гольца синдром) |
| 404 | Фокально-кортикальная дисплазия Тейлора |
| 405 | Фон Хиппель-Линдау Синдром |
| 406 | Фруктозо1,6 дифосфотазы недостаточность |
| 407 | Фукозидоз |
| 408 | Хайду-Чейни синдром |
| 409 | Хондродисплазия метафизарная тип Мак-Кьюсика |
| 410 | Хондродисплазия точечная Конради-Хюнермана |
| 411 | Хондрокальциноз |
| 412 | Хореоатетоз, гипотиреоидизм и неонатальная дыхательная недостаточность |
| 413 | Хорея Гентингтона |
| 414 | Хорея доброкачественная наследственная |
| 415 | Хороидермия |
| 416 | Хромосомные болезни |
| 417 | Хроническая гранулематозная болезнь |
| 418 | Х-сцепленная агаммаглобулинемия |
| 419 | Х-сцепленный лимфопролиферативный синдром (болезнь Дункана, синдром Пуртильо) |
| 420 | Х-сцепленный моторный нистагм |
| 421 | Х-сцепленный тяжелый комбинированный иммунодефицит |
| 422 | Целвегера синдром |
| 423 | Центронуклеарная миопатия |
| 424 | Цереброокулофациоскелетный синдром |
| 425 | Цистиноз |
| 426 | Цистиноз нефропатический |
| 427 | Цитруллинемия тип 1 |
| 428 | Шварца-Джампела синдром |
| 429 | Швахмана-Даймонда синдром |
| 430 | Шегрена-Ларссона синдром |
| 431 | Шерешевского-Тернера синдром |
| 432 | Широкого водопровода преддверия синдром |
| 433 | Шпринтцена-Гольдберга синдром |
| 434 | Штаргардта болезнь |
| 435 | Эдвардса синдром |
| 436 | Экзостозы множественные |
| 437 | Эксудитивная витреохореорстинальная дистрофия |
| 438 | Эктодермальная ангидротическая дисплазия |
| 439 | Эктодермальная гидротическая дисплазия |
| 440 | Эктопия хрусталика |
| 442 | Эллерса-Данло синдром |
| 443 | Эпилепсия прогрессирующая миоклоническая |
| 444 | Эпифизарная дисплазия, множественная |
| 445 | Эритрокератодермия |
| 446 | Эритроцитоз рецессивный |
| 447 | Эскобара cиндром |
Дистальный артрогрипоз 5-го типа – артрогрипоз с офтальмоплегией, полиневропатией.
 Клинический случай
Клинический случай
Представлен клинический случай пациентки 27 лет с дистальным артрогрипозом 5-го типа – артрогрипозом с офтальмоплегией, который сочетался у больной с полиневропатией. Для оценки тканевого дыхания (дыхательной цепи митохондрий) и других видов обмена в митохондриях проводили цитохимический анализ лимфоцитов в периферической крови по методу A. Pearse в модификации Р.П. Нарциссова. Оценивали активность 4 ферментов митохондрий, участвующих в углеводном обмене (лактатдегидрогеназа), обмене аминокислот (глутаматдегидрогеназа), обмене жирных кислот (α-глицерофосфатдегидрогеназа) и II комплексе дыхательной цепи митохондрий (сукцинатдегидрогеназа). Определено легкое снижение активности фермента сукцинатдегидрогеназы, входящей во второй комплекс дыхательной цепи митохондрий. Более значительно снижена активность фермента α-глицерофосфатдегидрогеназы, отмечено повышение активности лактатдегидрогеназы. В представленном наблюдении наряду с типичными проявлениями заболевания (контрактуры кистей и стоп, офтальмоплегия, птоз век, нарушение зрения) выявлена полиневропатия с нарушением чувствительности по полиневритическому типу. Таким образом, у больной с полиневропатией оказалось наследственное заболевание – артрогрипоз 5-го типа. Наряду с типичными проявлениями заболевания выявлена полиневропатия с гипорефлексией и нарушением чувствительности по полиневритическому типу. Были выявлены вторичные митохондриальные нарушения, что было основанием для назначения энерготропной терапии.
Таким образом, у больной с полиневропатией оказалось наследственное заболевание – артрогрипоз 5-го типа. Наряду с типичными проявлениями заболевания выявлена полиневропатия с гипорефлексией и нарушением чувствительности по полиневритическому типу. Были выявлены вторичные митохондриальные нарушения, что было основанием для назначения энерготропной терапии.
Ключевые слова: артрогрипоз, артрогрипоз 5-го типа, полиневропатия, митохондрии, дыхательная цепь, сукцинатдегидрогеназа, глицерофосфатдегидрогеназа, глутаматдегидрогеназа, лактатдегидрогеназа
________________________________________________
A clinical case of a 27-year-old patient with distal arthrogryposis of the 5th type – arthrogryposis with ophthalmoplegia, which was combined in a patient with polyneuropathy is presented. To assess tissue respiration (mitochondrial respiratory chain) and other types of metabolism in mitochondria, cytochemical analysis of lymphocytes in peripheral blood was carried out according to A. Pearse’s method modified by R.P. Narcissov. The activity of four mitochondrial enzymes involved in carbohydrate metabolism (lactate dehydrogenase), amino acid metabolism (glutamate dehydrogenase), fatty acid metabolism (α-glycerophosphate dehydrogenase) and the second complex of the mitochondrial respiratory chain (succinate dehydrogenase) was assessed. A slight decrease in the activity of the enzyme succinate dehydrogenase, which is part of the second complex of the mitochondrial respiratory chain, was determined. The activity of the enzyme α-glycerophosphate dehydrogenase was more significantly reduced, an increase in the activity of lactate dehydrogenase was noted. In the presented observation, along with the typical manifestations of the disease (contractures of the hands and feet, ophthalmoplegia, ptosis of the eyelids, visual impairment), polyneuropathy with impaired sensitivity of the polyneuritic type was revealed. Thus, a patient with polyneuropathy had a hereditary type 5 arthrogryposis disease.
Pearse’s method modified by R.P. Narcissov. The activity of four mitochondrial enzymes involved in carbohydrate metabolism (lactate dehydrogenase), amino acid metabolism (glutamate dehydrogenase), fatty acid metabolism (α-glycerophosphate dehydrogenase) and the second complex of the mitochondrial respiratory chain (succinate dehydrogenase) was assessed. A slight decrease in the activity of the enzyme succinate dehydrogenase, which is part of the second complex of the mitochondrial respiratory chain, was determined. The activity of the enzyme α-glycerophosphate dehydrogenase was more significantly reduced, an increase in the activity of lactate dehydrogenase was noted. In the presented observation, along with the typical manifestations of the disease (contractures of the hands and feet, ophthalmoplegia, ptosis of the eyelids, visual impairment), polyneuropathy with impaired sensitivity of the polyneuritic type was revealed. Thus, a patient with polyneuropathy had a hereditary type 5 arthrogryposis disease. Along with the typical manifestations of the disease, polyneuropathy with hyporeflexia and impaired sensitivity of the polyneuritic type was revealed. Secondary mitochondrial disorders were identified, which was the basis for the appointment of energotropic therapy.
Along with the typical manifestations of the disease, polyneuropathy with hyporeflexia and impaired sensitivity of the polyneuritic type was revealed. Secondary mitochondrial disorders were identified, which was the basis for the appointment of energotropic therapy.
Keywords: arthrogryposis, type 5 arthrogryposis, polyneuropathy, mitochondria, respiratory chain, succinate dehydrogenase, glycerophosphate dehydrogenase, glutamate dehydrogenase, lactate dehydrogenase
Детская ортопедия: артогрипоз лучше лечить на ранней стадии | 161.ru
Артрогрипоз – врожденное заболевание, характеризующееся контрактурами двух и более крупных суставов несмежных областей, а также поражением мышц и спинного мозга. Под термином «контрактура» подразумевается ограничение двигательной функции сустава.
Сейчас выявлено более 150 причин, вызывающих это заболевание: вирусные и бактериальные инфекции, физические факторы, химические вещества, лекарственные препараты, ограничение внутриматочного пространства (аномалии формы матки), плацентарная недостаточность, многоводие, и т.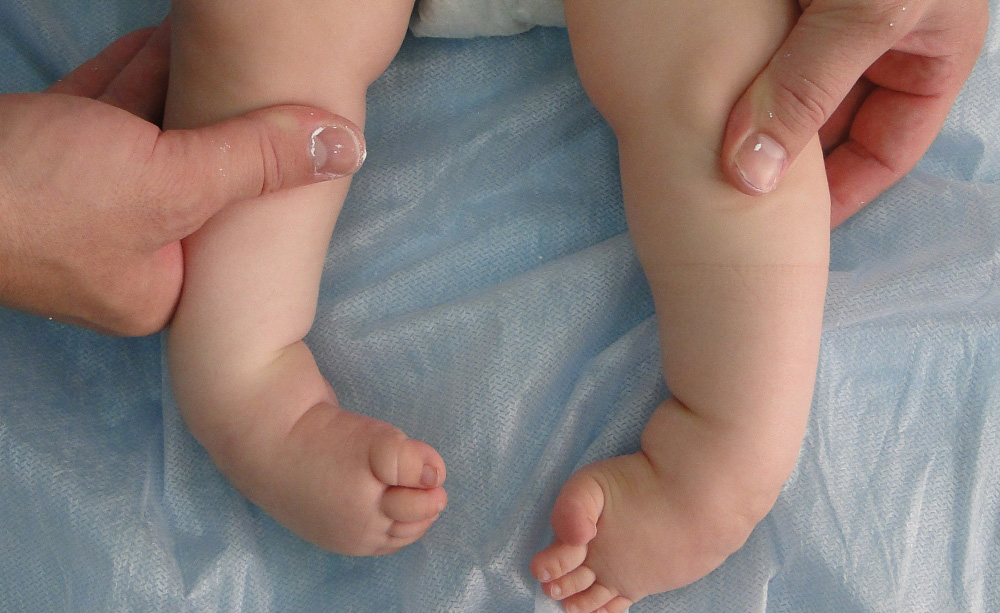 д. В анамнезе у матерей отмечаются различные заболевания, а также токсикоз беременности, выкидыши, аборты и пр. Причины развития артрогрипоза пока не известны.
д. В анамнезе у матерей отмечаются различные заболевания, а также токсикоз беременности, выкидыши, аборты и пр. Причины развития артрогрипоза пока не известны.
Воздействие тератогенного фактора на ранних сроках беременности вызывает нарушение развития мышечных волокон, или же приводит к первичному повреждению спинного мозга, что, в свою очередь, вызывает вторичные изменения мышц. При этом отмечается избирательный характер поражения мышц. В результате возникает дисбаланс в мышечном тонусе, что ограничивает движения в суставах, приводит к укорочению связок и других околосуставных тканей и клинически проявляется в виде фиксации сустава в определенном положении.
В настоящее время наличие патологии можно заподозрить еще во время беременности: отмечаются позднее шевеление плода, слабая его двигательная активность.
В большинстве случаев артрогрипоз не передается по наследству и проявляется как спорадический случай.
Различают следующие типы артрогрипоза: генерализованный (54%), с поражением нижних конечностей (30%), с поражением верхних конечностей (5%) и дистальный (11%).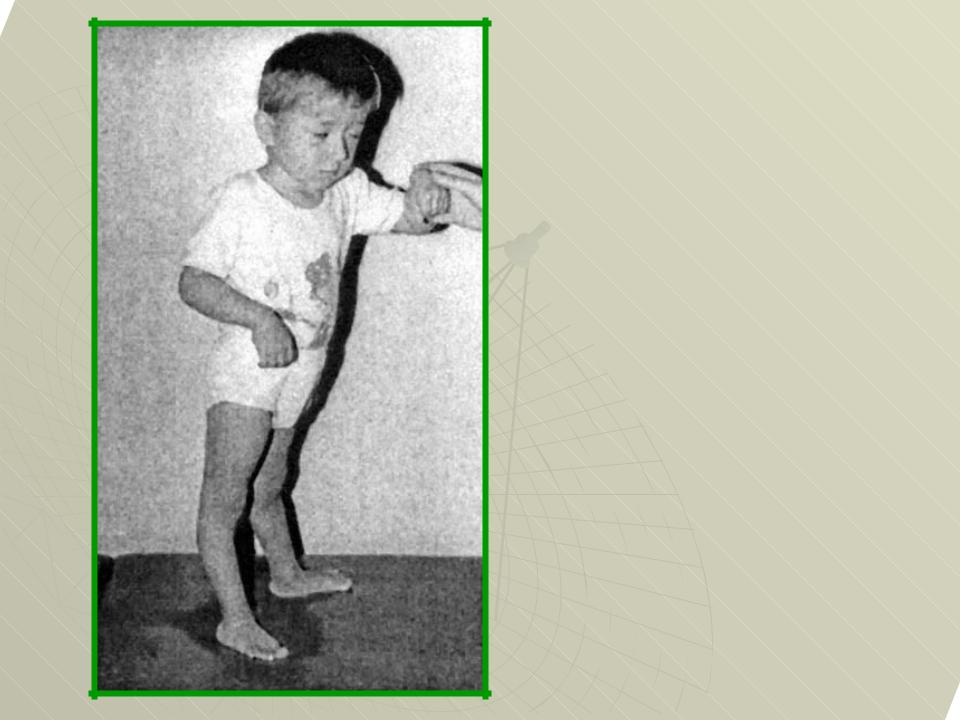
При генерализованнном типе артрогрипоза в тяжелых случаях отмечается поражение плечевых, локтевых, лучезапястных, тазобедренных, коленных суставов, деформации кистей и стоп, лицевого скелета. Возможны деформации позвоночника. Характерна мышечная гипотония или атония.
При дистальном типе артрогрипоза в большинстве случаев наблюдается деформации кистей и стоп, которые в некоторых случаях сочетаются с патологией крупных суставов конечностей. В подавляющем большинстве случаев деформации симметричные и не прогрессируют в процессе жизни ребенка.
Поражение внутренних органов, как правило, не наблюдается. Интеллект больных в подавляющем большинстве случаев сохранен, а в некоторых случаях такие дети опережают своих сверстников в интеллектуальном развитии. Своевременное начало лечения позволяет в дальнейшем обучаться в обычных школах и вести полноценный образ жизни.
Лечить артрогрипоз необходимо на ранних его стадиях. В лечении помогает активная помощь родителей, их терпение и желание добиться результата, несмотря на тяжесть деформаций.
С первых дней жизни больной должен быть осмотрен ортопедом. С 4-5-дневного возраста ребенку необходимо этапное гипсование для устранения деформаций стоп, коленных суставов. Для коррекции контрактур локтевых, лучезапястных суставов, пальцев кисти изготавливаются гипсовые лонгеты.
Родители обучаются корригирующим упражнениям и укладкам на устранение контрактур и деформаций в суставах верхних и нижних конечностей, которые выполняют по 6-8 раз в день. После занятий лечебной физкультурой (ЛФК) конечности фиксируются туторами или лонгетами. При достижении большего угла коррекции изготавливаются новые ортезные изделия.
С первых двух-трех недель, как только кожный покров ребенка будет адаптирован к механической нагрузке, детям, страдающим артрогрипозом, делается общий массаж. (курс – 15-20 сеансов; в год – 5-6 курсов).
ЛФК, массаж сочетаются с физиотерапевтическим лечением (солевые грелки, парафиновые или озокеритовые аппликации, электрофорез с препаратами, улучшающими проведение нервных импульсов и улучшающими микроциркуляцию тканей, магнитоимпульсная и электростимуляция, электрофорез с лидазой и т. д.), а также неврологическим (средства улучшающие проводимость, кровообращение и трофику тканей).
д.), а также неврологическим (средства улучшающие проводимость, кровообращение и трофику тканей).
Своевременное начало консервативного лечения позволяет в ряде случаев полностью устранить контрактуры или в значительной степени уменьшить их тяжесть.
При сохранении деформаций конечностей после проведенного консервативного лечения с 3-4 месячного возраста ребенка показано оперативное лечение.
В профилактике развития рецидивов контрактур важную роль играет ортезное снабжение больного (тутора, лонгеты, ортопедическая обувь, корректоры осанки, корсеты).
Вследствие чрезвычайной склонности артрогрипотических контрактур и деформаций к рецидиву необходимо длительное ношение фиксационных аппаратов и диспансерное наблюдение за больными до окончания их роста.
Прогноз при генерализованных формах артрогрипоза в отношении восстановления формы и функции сустава всегда серьезен, при локализованных формах – благоприятен. В результате комплексного и длительного лечения удается значительно улучшить статико-динамические возможности больного.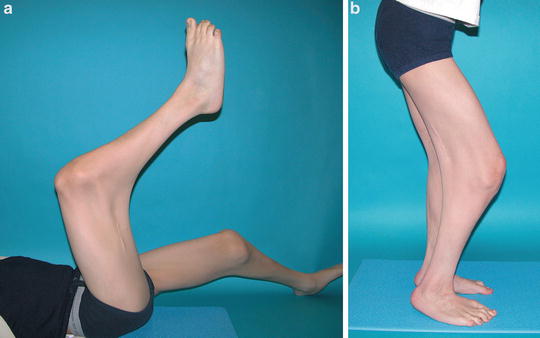
Консультативный прием завотделения в отделении детской ортопедии
Руководитель отделения детской травматологии и ортопедии для детей – главный детский ортопед Ростова-на-Дону, к.м.н. Мурадьян Владимир Юрьевич.
МБУЗ ГБ№20,
адрес: Коммунистический, 39, 4 этаж;
часы приема: по субботам, с 10:00 до 13:00;
контактный телефон: (863) 271-97-20.
Публикации в СМИ
Контрактура суставов — стойкое ограничение подвижности в суставе.
Классификация • По происхождению: •• Врождённая •• Приобретённая • По этиологии: •• Артрогенная — при патологии суставных поверхностей сочленяющихся костей, связок и капсулы сустава •• Болевая (анталгическая) — рефлекторное ограничение движений в суставе при болезненности движений •• Дерматогенная — при обширных рубцовых изменениях кожи •• Десмогенная — при рубцовых изменениях соединительнотканных образований (фасций, апоневрозов и др. ) •• Миогенная — при укорочении мышц в результате травмы, воспалительных или дистрофических процессов •• Неврогенная — при нарушениях иннервации •• Паралитическая — при параличе мышцы или группы мышц •• Послеампутационная контрактура — осложнение ампутации конечности в виде контрактуры ближайшего к культе сустава; развивается при неправильной технике операции или при погрешностях послеоперационного ведения •• Профессиональная — контрактура при хронической травматизации или перенапряжении определённых групп мышц в связи с профессиональной деятельностью •• Психогенная (истерическая) — неврогенная контрактура при истерии •• Рефлекторная — контрактура при длительном раздражении нерва, приводящего к возникновению стойкого рефлекса в виде повышения тонуса мышцы или группы мышц •• Рубцовая — контрактура при грубых рубцовых изменениях тканей •• Спастическая — контрактура при центральном параличе (парезе) •• Сухожильная (тендогенная) — контрактура при укорочении сухожилия •• Функционально-приспособительная (компенсаторная) — контрактура, развивающаяся для компенсации анатомического дефекта, например сгибательная контрактура суставов одной ноги при укорочении другой • По характеру: •• Разгибательная — контрактура с ограничением сгибания в суставе •• Сгибательная — контрактура с ограничением разгибания в суставе.
) •• Миогенная — при укорочении мышц в результате травмы, воспалительных или дистрофических процессов •• Неврогенная — при нарушениях иннервации •• Паралитическая — при параличе мышцы или группы мышц •• Послеампутационная контрактура — осложнение ампутации конечности в виде контрактуры ближайшего к культе сустава; развивается при неправильной технике операции или при погрешностях послеоперационного ведения •• Профессиональная — контрактура при хронической травматизации или перенапряжении определённых групп мышц в связи с профессиональной деятельностью •• Психогенная (истерическая) — неврогенная контрактура при истерии •• Рефлекторная — контрактура при длительном раздражении нерва, приводящего к возникновению стойкого рефлекса в виде повышения тонуса мышцы или группы мышц •• Рубцовая — контрактура при грубых рубцовых изменениях тканей •• Спастическая — контрактура при центральном параличе (парезе) •• Сухожильная (тендогенная) — контрактура при укорочении сухожилия •• Функционально-приспособительная (компенсаторная) — контрактура, развивающаяся для компенсации анатомического дефекта, например сгибательная контрактура суставов одной ноги при укорочении другой • По характеру: •• Разгибательная — контрактура с ограничением сгибания в суставе •• Сгибательная — контрактура с ограничением разгибания в суставе.
Лечение • Раннее и комплексное • Лечение основного заболевания • ЛФК, физиотерапия (электрофорез с лидазой, ронидазой, фонофорез с гидрокортизоном, динатриевой солью этилендиаминтетрауксусной кислоты), массаж • При артрогенных контрактурах — внутрисуставные гидравлические новокаиновые блокады • При безуспешности консервативного лечения — оперативное (артролиз, пластические операции и др.).
Профилактика — пассивная и активная ранняя лечебная гимнастика при заболеваниях, приводящих к формированию контрактур.
МКБ-10 • M24.5 Контрактура сустава.
Приложение. Артрогрипоз — врождённые множественные контрактуры вследствие недоразвития мышц конечностей. Различают несколько генетических разновидностей, в частности аутосомно-доминантные формы (*108110; 108120 [9p22–9q22.3, дефект гена AMCD1] — артрогрипоз множественный врождённый, дистальный, тип 1; 108130; 108140; 108145; 108200), аутосомно-рецессивные (*208080; 208081; 208100 [5q35, дефект гена AMCN1]; 208110; 208150; 208200) и X-сцепленные (*301820; 301830 [Xp11. 3–Xq11.2, дефект гена AMCD] — артрогрипоз множественный врождённый, дистальный)
3–Xq11.2, дефект гена AMCD] — артрогрипоз множественный врождённый, дистальный) « Амиоплазия врождённая « Артромиодисплазия врождённая. МКБ-10 • Q74.3 Врождённый множественный артрогрипоз
Мультиплексный конгенита артрогрипоза — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Hall JG. Артрогрипозы (множественные врожденные контрактуры). В: Принципы и практика медицинской генетики Эмери и Римоина, 6-е изд. Римоин Д.Л., Коннор Дж. М., Пайериц Р. Е., Корф Б. Р., ред., Черчилль Ливингстон: Нью-Йорк, 2012 г.
Холл Дж. Дж. Артрогрипоз. В: Управление генетическими синдромами, 3-е изд. Кэссиди С.Б., Аллансон Дж. Э. ред., Джон Вили и сыновья: Хобокен, Нью-Джерси, 2010; 81-96.
Джонс KL. Эд. Распознаваемые модели пороков развития человека Смита. 6-е изд. Эльзевьер Сондерс, Филадельфия, Пенсильвания; 2006: 774-777.
Бамшад М. Врожденный множественный артрогрипоз. Руководство NORD по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 155.
Руководство NORD по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 155.
Берков Р., изд. Руководство Merck — домашнее издание, 2-е изд. Станция Уайтхаус, Нью-Джерси: Исследовательские лаборатории Мерк; 2003: 1524-1525.
Staheli L T, Hall JG, Jaffe K M, Paholke DO. Артрогрипоз: текстовый атлас.Издательство Кембриджского университета; Кембридж, Великобритания, 1998 г.
СТАТЬИ ЖУРНАЛА
Исправление в: Am J Med Genet 2015; 167A: 2866. Огранович, Алга [исправлено на Огранович, Алга]; Понтен, Ава [исправлено на Понтен, Ева].
Hall JG, Agranovich O, Pontén E, van Bosse HJ. Резюме 2-го Международного симпозиума по артрогрипозу, Санкт-Петербург, Россия, 17-19 сентября 2014 г. Am J Med Genet 2015; 167A: 1193-1195.
Холл, JG. Последовательность олигогидрамниона пересмотрена в связи с артрогрипозом с характерными кожными изменениями.Am J Med Genet 2014; 164A: 2775-2792.
Hall JG, Aldinger KA, Tanaka, KI. Возвращение к амиоплазии. Am J Med Genet 2014; 164: 700-730.
Am J Med Genet 2014; 164: 700-730.
Холл JG. Артрогрипоз (множественные врожденные контракты): диагностический подход к этиологии, классификации, генетике и общим принципам. Eur J Med Genet 2014; 57: 464-472.
Filges I, зал JG. Неспособность идентифицировать антенатальные множественные врожденные контрактуры и акинезию плода — предложение рекомендаций по улучшению диагностики. Prenat Diag 2013; 33: 61-74.
Холл JG. Структурные аномалии матки и артрогрипоз — смерть городской легенды. Am J Med Genet 161A 2012; 82-88.
Лоури РБ, Сиббальд Б., Бедард Т, Холл Дж. Дж. Распространенность множественных врожденных контрактур, включая врожденный множественный артрогрипоз, в Альберте, Канада, и стратегия классификации и кодирования. Врожденные дефекты Res A Clin Mol Teratol 2010; 88: 1057-1061.
Диллон Э.Р., Бьорнсон К.Ф., Джаффе К.М., Холл Дж. Г., Сонг К. Амбулаторная активность у молодежи с артрогрипозом: когортное исследование.J Pediatr Orthop. 2009; 29: 214-217.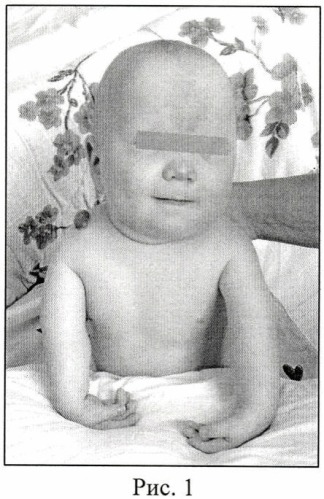
Холл JG. Возвращение к фенотипу Pena Shokeir (последовательность деформации акинезии плода). Врожденные дефекты Res A, 2009; 85: 677-694.
Бамшад М., ван Хест А.Е., Удовольствие Д. Артрогрипоз: обзор и обновление. J Bone Joint Surg Am. 2009; 91: 40-46.
Fassier A, Wicart P, Dubousset J, Seringe R. Врожденный множественный артрогрипоз. Долгосрочное наблюдение от рождения до формирования скелета. J Детский Ортоп. 2009; 3: 383-390.
Беван В.П., Холл Дж. Г., Бамшад М. и др.Врожденный множественный артрогрипоз (амиоплазия): ортопедическая перспектива. J Pediatr Orthop. 2007; 27: 594-600.
Бернштейн РМ. Артрогрипоз и амиоплазия. J Am Acad Orthop Surg. 2002; 10: 417-424.
Продает JM, Jaffe KM, Hall JG. Амиоплазия, наиболее распространенный тип артрогрипоза: возможность хорошего результата. Педиатрия. 1996; 97: 225-231.
ИНТЕРНЕТ
Лал МК. Артрогрипоз. Medscape. Обновлено: 3 января 2019 г. Доступно по адресу: http://emedicine.medscape. com/article/941917-overview Проверено 2 мая 2019 г.
com/article/941917-overview Проверено 2 мая 2019 г.
Информационный центр по генетическим и редким заболеваниям (GARD). Врожденный множественный артрогрипоз (AMC). Последнее обновление: 12.01.2015. Доступно по адресу: http://rarediseases.info.nih.gov/GARD/Disease.aspx?PageID=4&diseaseID=777 По состоянию на 2 мая 2019 г.
Дистальный артрогрипоз: новый тип с четко выраженным лицом и отсутствием зубов
Редактор. Дистальные артрогрипозы — это группа наследственных заболеваний с врожденными контрактурами дистальных отделов конечностей. Отец и дочь с похожими контрактурами дистальных отделов конечностей, отчетливыми чертами лица, вдавленными коронковыми швами и врожденным отсутствием зубов характеризуют новый тип дистального артрогрипоза.
Отчет о болезни
Девочка 11 лет была осмотрена для оценки контрактур пальцев. При рождении у нее были отмечены контрактуры пальцев и «опухшее лицо» (рис. 1). Ее вес при рождении был 4077 г, а длина — 51 см. В 11 месяцев у нее появились «спусковые крючки» пятых пальцев с некоторым улучшением. В возрасте девяти лет ей диагностировали синдром дефицита внимания и лечили риталином. В 10 лет лечили от контрактуры и уравновешивали сухожилие мизинца правой руки.На мизинец левой руки наложили шину. Клинических данных о неврологических или мышечных заболеваниях не было. Она была на уровне 50-го центиля до 9 лет, когда скорость роста снизилась.
В возрасте девяти лет ей диагностировали синдром дефицита внимания и лечили риталином. В 10 лет лечили от контрактуры и уравновешивали сухожилие мизинца правой руки.На мизинец левой руки наложили шину. Клинических данных о неврологических или мышечных заболеваниях не было. Она была на уровне 50-го центиля до 9 лет, когда скорость роста снизилась.
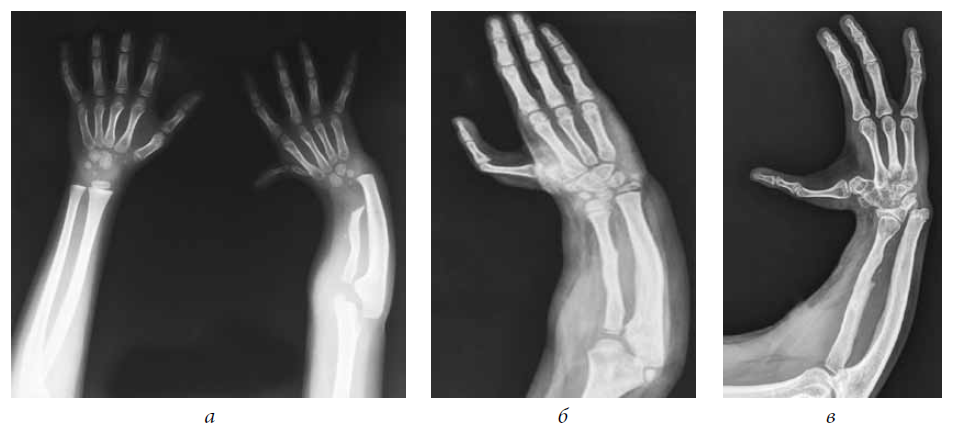
фигура 1
AP фотографии пробанда при рождении (A), в возрасте 11 лет (B), и ее отца (C), на которых видны маленькие горизонтальные глазные щели, широкое основание носа и форма верхней губы в форме лука купидона. Вдавление коронарной щели ощутимо, но не видно на фотографии.
Осмотр через 11 лет показал окружность головы 51 см (50%), небольшие горизонтальные глазные щели, эпикантические складки, легкую микрогнатию и пальпируемое углубление коронкового поперечного шва. Нос расширился, прилегая к эпикантической складке, колумелла находилась под кончиком носа, а верхняя губа имела форму лука купидона (рис. 1). Восемь постоянных двустворчатых зубов и верхний левый боковой резец отсутствовали врожденно (рис. 2). На руках были видны выступающие подушечки суставов пальцев, симметричные легкие контрактуры проксимальных межфаланговых суставов указательного, среднего и безымянного пальцев и 45 ° контрактура проксимального межфалангового сустава мизинцев обеих рук.Большие пальцы рук сведены и согнуты. Присутствовала поперечная ладонная складка, а сгибательные складки пальцев не были хорошо развиты (рис. 3). Локти и запястья слегка не разгибались. Пронация и супинация в норме. Диапазон движений плеч и бедер был нормальным. Контрактура коленей 10 °. Ступни показали умеренную жесткость пальцев без деформации. Зрение, слух, речь, интеллект, моторное развитие и хромосомные исследования были нормальными. Контрактуры коленного сустава и контрактуры пальцев лечили ночными шинами без каких-либо улучшений.
2). На руках были видны выступающие подушечки суставов пальцев, симметричные легкие контрактуры проксимальных межфаланговых суставов указательного, среднего и безымянного пальцев и 45 ° контрактура проксимального межфалангового сустава мизинцев обеих рук.Большие пальцы рук сведены и согнуты. Присутствовала поперечная ладонная складка, а сгибательные складки пальцев не были хорошо развиты (рис. 3). Локти и запястья слегка не разгибались. Пронация и супинация в норме. Диапазон движений плеч и бедер был нормальным. Контрактура коленей 10 °. Ступни показали умеренную жесткость пальцев без деформации. Зрение, слух, речь, интеллект, моторное развитие и хромосомные исследования были нормальными. Контрактуры коленного сустава и контрактуры пальцев лечили ночными шинами без каких-либо улучшений.
фигура 2
Рентгенограмма челюсти пробанда AP показывает врожденное отсутствие двустворчатых зубов.
Рисунок 3
На фотографии
AP руки пробанда 11 лет (A) и ее отца (B) показаны контрактуры пальцев, сведенные и согнутые большие пальцы, поперечная ладонная складка и недоразвитие сгибательных складок суставов пальцев.
В 12 лет ее рост составлял 142 см, а вес — 31 кг. Из-за постепенного снижения достигнутого роста ее обследовали с помощью тестов на стимуляцию инсулина и аргинина, которые показали дефицит гормона роста.Ее лечили гормоном роста человека, и рост ее скелета увеличился с 29-го центиля в возрасте 12 лет до 50-го центиля.
В возрасте 14 лет у нее была замечена контрактура сгибания 10 ° левого локтя и контрактуры колена 10 ° и 15 °. Контрактуры коленного сустава лечили гипсованием с минимальным улучшением. В возрасте 15 лет диапазон пассивных движений проксимальных межфаланговых и дистальных межфаланговых суставов безымянного и мизинца составлял от 35 ° сгибания до 50 ° сгибания.Большие пальцы имели полный диапазон пассивных движений. В возрасте 16 лет прогрессирующая деформация бурсита правой стопы была вылечена плюсневой остеотомией и рифлением медиальной капсулы.
Семья включает отца, пострадавшего так же, и мать и сестру, не пострадавшие от болезни. Ее отец был ростом 1,72 м, а черты лица были такими же, как у его дочери (рис. 1). У него врожденное отсутствие восьми зубов, сейчас носит зубные протезы. На его руках были обнаружены легкие контрактуры проксимальных межфаланговых суставов пальцев и выступающие подушечки суставов.Сгибание дистальных межфаланговых суставов было ограничено 45 °, а сгибательные складки дистальных межфаланговых суставов были недоразвиты (рис. 3). Его большие пальцы были сведены и согнуты, но фиксированных контрактур не было. Проксимальные межфаланговые суставы пальцев стопы и межфаланговые суставы большого пальца стопы жесткие. Коленных контрактур не было. Его правому локтю не хватало 15 ° разгибания, а предплечью — 10 ° пронации. У него диагностировали синдром дефицита внимания, болезнь Меньера и отосклероз.
1). У него врожденное отсутствие восьми зубов, сейчас носит зубные протезы. На его руках были обнаружены легкие контрактуры проксимальных межфаланговых суставов пальцев и выступающие подушечки суставов.Сгибание дистальных межфаланговых суставов было ограничено 45 °, а сгибательные складки дистальных межфаланговых суставов были недоразвиты (рис. 3). Его большие пальцы были сведены и согнуты, но фиксированных контрактур не было. Проксимальные межфаланговые суставы пальцев стопы и межфаланговые суставы большого пальца стопы жесткие. Коленных контрактур не было. Его правому локтю не хватало 15 ° разгибания, а предплечью — 10 ° пронации. У него диагностировали синдром дефицита внимания, болезнь Меньера и отосклероз.
Обсуждение
Артрогрипоз определяется как наличие непрогрессирующих врожденных контрактур двух или более участков тела. Дистальный артрогрипоз в первую очередь поражает дистальные отделы конечностей и был классифицирован Hall et al .
1 в 1982 г. Классификация Холла различала шесть типов периферических контрактур, основанных на наличии или отсутствии сопутствующих аномалий в дополнение к типу периферических контрактур. Он не включал все синдромы, о которых известно, что они имеют периферические контрактуры, и включал некоторые, о которых было известно, что они не наследуются.1 Классификация Холла была изменена Бамшадом и др.
Он не включал все синдромы, о которых известно, что они имеют периферические контрактуры, и включал некоторые, о которых было известно, что они не наследуются.1 Классификация Холла была изменена Бамшадом и др.
2, которые определили дистальный артрогрипоз как «наследственное первичное уродство конечностей, характеризующееся врожденными контрактурами двух или более различных участков тела и без первичного неврологического и / или мышечного заболевания, которое влияет на функцию конечностей». Деформации верхних конечностей включают врожденное отклонение локтевой кости, камптодактилию (или псевдокамптодактилию), гипопластические и / или отсутствующие сгибательные складки и перекрытие пальцев.Деформации нижних конечностей включают эквиноварусную косолапость, пяточно-вальгусную, вертикальную таранную кость и варусную плюсну, но не ограничиваются этими деформациями. Было описано девять синдромов пороков развития, отвечающих этим критериям, которые имеют общую картину контрактуры дистального сустава, минимальное поражение проксимального сустава и аутосомно-доминантное наследование. 1-11 Хотя классификация является клинически полезной, некоторые из этих состояний, как было показано, имели место. генетическая гетерогенность.12
1-11 Хотя классификация является клинически полезной, некоторые из этих состояний, как было показано, имели место. генетическая гетерогенность.12
Описанная здесь семья отличается от других пациентов с дистальным артрогрипозом.У этой семьи врожденные контрактуры пальцев и деформация большого пальца. Отличительные черты лица присутствовали при рождении и включали небольшие и горизонтальные глазные щели, расширение носа, форму верхней губы купидона, углубление коронарного шва и врожденное отсутствие двустворчатых зубов. Легкие контрактуры в локтях и коленях и ограниченное движение пальцев ног. Лечение контрактур колена и пальцев у этого пациента имело ограниченный успех.Эти черты есть у отца и дочери. Неясно, является ли дефицит гормона роста, присутствующий у основного пациента, частью синдрома. Для выяснения фенотипа и наследования этого синдрома потребуется отчет о других аналогичных семьях.
Артрогрипоз | Общая информация — Группа артрогрипоза
Мультиплексный конгенита артрогрипоза (AMC) — это термин, используемый для описания более 300 состояний, вызывающих множественные искривления суставов на разных участках тела при рождении.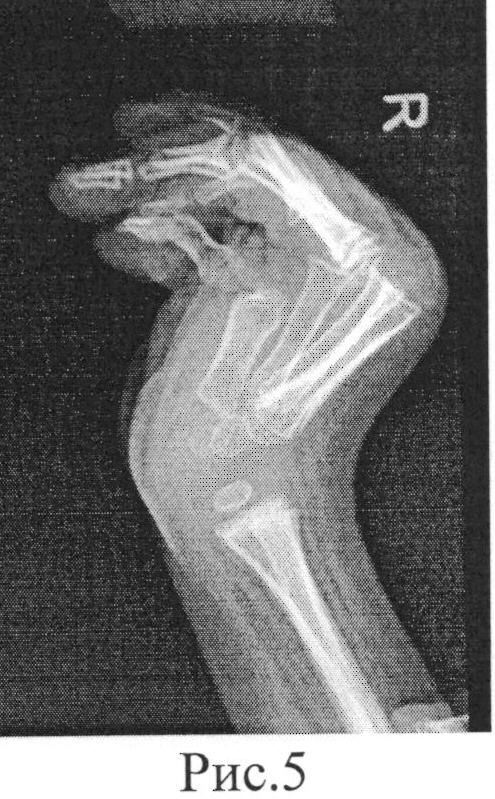 Он варьируется от человека к человеку, обычно жесткие суставы и мышечная слабость.
Он варьируется от человека к человеку, обычно жесткие суставы и мышечная слабость.
AMC — это не конкретный диагноз, а, скорее, клинический результат постоянного укорочения суставов, также называемый непрогрессирующими врожденными контрактурами.
AMC является непрогрессивным, что означает, что отсутствие движения не ухудшается с течением времени, однако людям рекомендуется обратиться за советом, чтобы потенциально предотвратить дальнейшие нарушения суставов.
Иногда AMC также может влиять на центральную нервную систему, выживаемость которой может быть низкой.
Arthrogryposis Multiplex Congenita (AMC) не является результатом проблемы с формированием суставов, а скорее является результатом развития соединительных тканей вокруг них, которое происходит после 8-10 недель беременности.
В AMC эта ткань соединяет сустав на месте, сильно ограничивая движение в пораженных областях, что приводит к тому, что сухожилия вокруг пораженного сустава не могут растянуться до своей нормальной длины. Ограничение движения в течение нескольких месяцев также может привести к контрактурам суставов.
Ограничение движения в течение нескольких месяцев также может привести к контрактурам суставов.
Существует множество причин конгенитационного множественного артрогрипоза (AMC), некоторые из которых передаются по наследству. Это:
Патологии соединительной ткани в области сухожилий, костей, суставов или выстилки суставов развиваются таким образом, что нормальные движения в матке невозможны.
Ограниченное пространство или ограниченное движение в матке , например, при многоплодных родах. В других случаях может быть недостаток нормального уровня околоплодных вод или у матери может быть аномальная форма матки, которая не позволяет ребенку свободно двигаться.
Нарушения структуры или функции мышц, также известные как миопатические процессы , при которых мышцы не формируются или развиваются, но не функционируют должным образом.
Аномалии нервов, которые соединяются с мышцами, также известные как невропатические процессы , когда нервы не могут формироваться, созревать или функционировать должным образом, что приводит к очень серьезному отсутствию движения и часто сопровождает структурные аномалии.
Сосудистая недостаточность, ведущая к потере нейронов — это проблема с нормальным кровообращением, которое затем не может питать нервы, ведущие к мышцам или костям, составляющим сустав.
Материнское заболевание — ряд материнских нарушений обмена веществ и материнских заболеваний были связаны с наличием множественных врожденных контрактур у ребенка.
Существует более 300 состояний, которые подпадают под «зонтик» артрогрипоза, и их можно разделить на три группы:
- Заболевания преимущественно конечностей
- заболевания конечностей и некоторых других участков тела, например волчья пасть, сердце, дефекты кишечника, искривление позвоночника
- Заболевания конечностей и центральной нервной системы
В основном конечность
Амиоплазия — наиболее распространенный тип заболевания, известный как «классический артрогрипоз».Это происходит у 1 из каждых 10 000 живорождений и составляет одну треть всех случаев людей, у которых диагностирован артрогрипоз. Обычно у людей поражаются все четыре конечности, но бывают случаи, когда поражаются только ноги или руки человека. Около 10% людей с амиоплазией имеют какой-либо тип аномалии кишечника или брюшной стенки, но это хорошо поддается ранней физиотерапии.
Обычно у людей поражаются все четыре конечности, но бывают случаи, когда поражаются только ноги или руки человека. Около 10% людей с амиоплазией имеют какой-либо тип аномалии кишечника или брюшной стенки, но это хорошо поддается ранней физиотерапии.
Это не считается генетическим заболеванием, но родителям ребенка с амиоплазией рекомендуется обратиться за генетической консультацией для своего ребенка, когда он станет взрослым.
При дистальном артрогрипозе I типа обычно наиболее сильно поражаются кисти и стопы. У новорожденного ребенка характеристики расстройства можно четко увидеть, когда руки сжаты, а пальцы перекрываются. Ступни также могут быть поражены вместе с коленями и бедрами, но обычно они довольно легкие. Опять же, у него относительно хороший ответ на раннюю физиотерапию.
Этот тип имеет аутосомно-доминантное наследование, и, поскольку только один родитель должен иметь аномальный ген, чтобы ребенок унаследовал болезнь, вероятность передачи его составляет 50/50.
Конечности и другие части тела
Множественные синдромы птеригиума
Птеригиум — это крыловидная структура, перепонка или треугольная мембрана, образующаяся поперек сустава тела. Существует много типов синдрома птеригиума, но у людей с этими состояниями часто бывают кожные перепонки на шее, коленях и локтях, а также множественные врожденные контрактуры. По краю этой паутины проходят кровеносные сосуды и нервы, поэтому во время операции требуется особая осторожность.
Каждый из различных синдромов птеригиума имеет разные формы наследования.
Синдром Фримена Шелдона (синдром свистящего лица)
Помимо контрактур кистей и стоп, это заболевание также имеет поражение лица. Мышцы сокращаются таким образом, что возникает «свистящий» вид. В самых тяжелых случаях движения рта могут быть крайне ограниченными. Даже если лицо не так поражено, человеку все равно может быть трудно улыбнуться, и в этой группе часто наблюдается постнатальный дефицит роста.
Дистальный артрогрипоз, тип II B
Этот тип DA также имеет признаки паралича всего или части глаза, и веки могут выглядеть опущенными.Помимо мышечной слабости вокруг глаз, сами глаза могут иметь ограниченное движение, особенно в стороны. Кожа и мышцы жесткие, на пальцах отсутствуют складки.
Этот тип аутосомно-доминантный и довольно распространенный.
Дистальный артрогрипоз, тип II C
У этого типа DA также есть заячья губа.
Дистальный артрогрипоз типа II D
При этом типе ДА также встречается сколиоз (искривление позвоночника).
Дистальный артрогрипоз типа II E
Этот тип DA имеет характерное положение руки, при котором запястье согнуто, но пястно-фаланговый сустав (ладонь к запястью) разогнут. Ограниченное раскрытие челюсти (тризм).
DA Type IIE относительно распространен и не передается по семействам.
Конечности плюс центральная нервная система
Младенцы из этой категории, как правило, плохо себя чувствуют и могут не выжить. К сожалению, это обычно совершенно очевидно в неонатальном периоде (первые 28 дней) и не развивается на более поздней стадии.
К сожалению, это обычно совершенно очевидно в неонатальном периоде (первые 28 дней) и не развивается на более поздней стадии.
Артрогрипоз — это разнообразное заболевание, и у него нет двух одинаковых людей. Однако благодаря физиотерапии, выборочному использованию хирургических вмешательств и ортезов (шин или штангенциркулей) большинство детей продолжают вести полноценный и активный образ жизни.
Для каждого ребенка, рожденного с артрогрипозом, важно как можно скорее поставить точный диагноз. Конкретный диагноз предоставит информацию о прогнозе, риске рецидива, а также подскажет практикующим врачам лучшие методы лечения.Чтобы помочь различать разные типы артрогрипоза, врачи, вероятно, будут использовать все доступные диагностические инструменты и, вероятно, спросят вас о:
Семейная история
Это важно, особенно если в семье есть другие больные дети. Врач изучит возраст отца и матери и спросит о близких браках, известных как кровное родство, в семье.
Пренатальный анамнез
Были ли у матери заболевания или лихорадка, травмы или травмы, какое-либо воздействие наркотиков, алкоголя, лекарств, которые могут вызвать врожденные дефекты? Врачи также будут специально спрашивать о движениях ребенка перед родами.
История рождений
Это будет включать продолжительность беременности и родов, время и продолжительность родов, положение ребенка при рождении и пренатальный исход.
Ребенку предстоит пройти ряд обследований для устранения других явных заболеваний, которые могут включать рентген позвоночника, таза и конечностей; биопсия мышц; МРТ; сканирование головы, а также исследования центральной нервной системы.
Физиотерапия
Невозможно переоценить успех коррекции положения суставов и конечностей с помощью физиотерапии.В течение первых 3-4 месяцев жизни, когда контрактуры имеют тенденцию к значительному ослаблению — известному как «период отсрочки» — некоторые мышцы, которые кажутся необычно слабыми, могут значительно увеличиваться в силе, а кости имеют тенденцию к увеличению минерализации и роста
Физиотерапию обычно можно начинать даже до того, как будет установлен конкретный диагноз. Вводя раннюю программу пассивного растяжения, пока ткани ребенка еще эластичны, это поможет увеличить диапазон движений в жестких суставах.Это часто сочетается с использованием шин для сохранения правильного положения конечности. Родителей следует обучать правильным методам лечения, чтобы они могли продолжать терапию дома между визитами в больницу. Часто использование серийных пластырей и корректирующих операций в более позднем возрасте дополняют эту работу и могут помочь ребенку стоять и ходить.
Вводя раннюю программу пассивного растяжения, пока ткани ребенка еще эластичны, это поможет увеличить диапазон движений в жестких суставах.Это часто сочетается с использованием шин для сохранения правильного положения конечности. Родителей следует обучать правильным методам лечения, чтобы они могли продолжать терапию дома между визитами в больницу. Часто использование серийных пластырей и корректирующих операций в более позднем возрасте дополняют эту работу и могут помочь ребенку стоять и ходить.
Трюковые движения, которые они изучают сами, часто могут помочь детям преодолеть трудности, которые могут возникнуть с их верхними конечностями. Если потребуется индивидуальное оборудование, терапевт сможет дать вам помощь и совет.
Большинство детей с артрогрипозом достигают некоторой степени подвижности; либо с помощью штангенциркуля, шины и костылей, либо полностью самостоятельно.
Загрузите более подробные советы по физиотерапии для детей с артрогрипозом от Миа Данкли, старшего педиатрического физиотерапевта в больнице Грейт-Ормонд-стрит, Лондон.
Дистальный артрогрипоз с вариабельной клинической экспрессией, вызванный мутацией TNNI2
Bamshad M, Van Heest AE, Pleasure D.Артрогрипоз: обзор и обновление. J Bone Joint Surg Am 2009; 91 : 40–46.
Артикул
Google Scholar
Wang B, Zheng Z, Wang Z, Zhang X, Yang H, Cai H et al. Новая миссенс-мутация TNNI2 в китайской семье вызывает дистальный артрогрипоз типа 1. Am J Med Genet A 2016; 170 : 135–141.
CAS
Статья
Google Scholar
Sung SS, Brassington AM, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA et al.Мутации в генах, кодирующих быстро сокращающиеся сократительные белки, вызывают синдромы дистального артрогрипоза. Am J Hum Genet 2003; 72 : 681–690.
CAS
Статья
Google Scholar
Sung SS, Brassington AM, Krakowiak PA, Carey JC, Jorde LB, Bamshad M.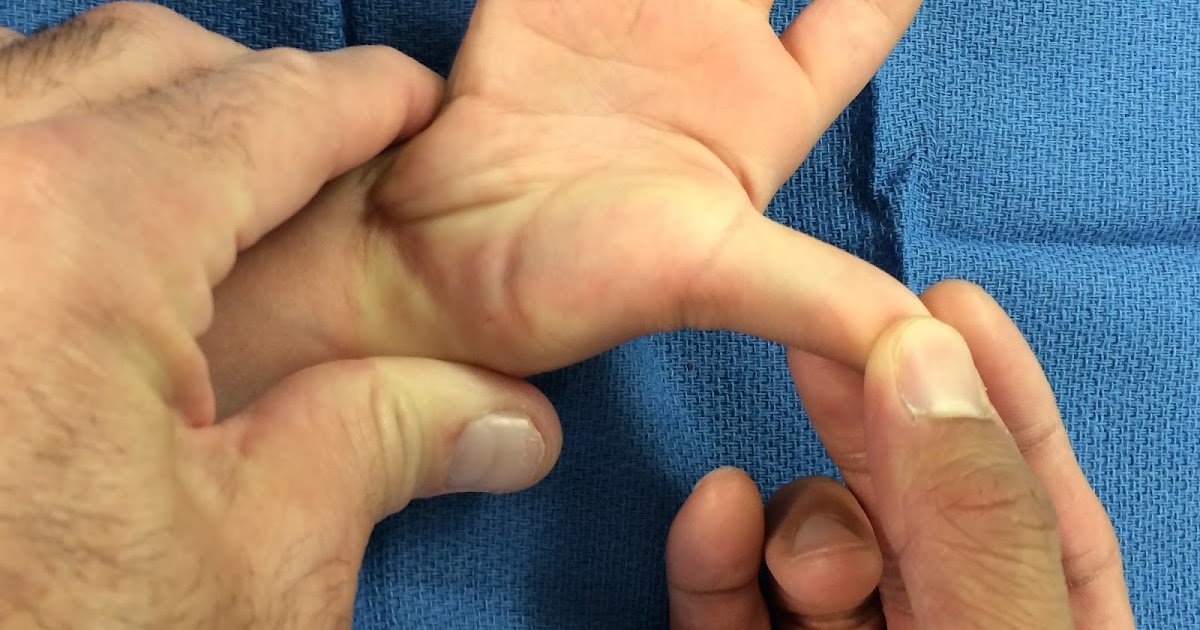 Мутации TNNT3 вызывают множественные врожденные контрактуры: второй локус для дистального артрогрипоза типа 2B. Am J Hum Genet 2003; 73 : 212–214.
Мутации TNNT3 вызывают множественные врожденные контрактуры: второй локус для дистального артрогрипоза типа 2B. Am J Hum Genet 2003; 73 : 212–214.
Артикул
Google Scholar
Beck AE, McMillin MJ, Gildersleeve HI, Kezele PR, Shively KM, Carey JC et al. Спектр мутаций, вызывающих дистальный артрогрипоз 1 и 2B типов. Am J Med Genet A 2013; 161A : 550–555.
Артикул
Google Scholar
Накашима М., Такано К., Цуюсаки Ю., Ёситоми С., Шимоно М., Аоки Ю. и др.Мутации WDR45 у трех пациентов мужского пола с синдромом Веста. J Hum Genet 2016; 61 : 653–661.
CAS
Статья
Google Scholar
Jiang M, Zhao X, Han W, Bian C, Li X, Wang G et al. Новая делеция TNNI2 вызывает дистальный артрогрипоз в большой китайской семье с заметной вариабельностью экспрессии. Hum Genet 2006; 120 : 238–242.
Hum Genet 2006; 120 : 238–242.
CAS
Статья
Google Scholar
Carli D, Fairplay T, Ferrari P, Sartini S, Lando M, Garagnani L et al.Генетические основы врожденных аномалий верхних конечностей: анализ 487 случаев специализированной клиники. Врожденные дефекты Res A Clin Mol Teratol 2013; 97 : 798–805.
CAS
Статья
Google Scholar
Shrimpton AE, Hoo JJ. Мутация TNNI2 в семье с дистальным артрогрипозом типа 2B. Eur J Med Genet 2006; 49 : 201–206.
Артикул
Google Scholar
Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M.Дистальный артрогрипоз: клинико-генетические данные. Acta Paediatr 2012; 101 : 877–887.
Артикул
Google Scholar
Парри Д. А., Сквайр Дж. М.. Структурная роль тропомиозина в регуляции мышц: анализ дифрактограмм расслабленных и сокращающихся мышц. J Mol Biol 1973; 75 : 33–55.
А., Сквайр Дж. М.. Структурная роль тропомиозина в регуляции мышц: анализ дифрактограмм расслабленных и сокращающихся мышц. J Mol Biol 1973; 75 : 33–55.
CAS
Статья
Google Scholar
Рарик Х.М., Ту XH, Соларо Р.Дж., Мартин А.Ф.С-конец сердечного тропонина I необходим для полной ингибирующей активности и чувствительности к Са2 + миофибрилл крыс. J Biol Chem 1997; 272 : 26887–26892.
CAS
Статья
Google Scholar
Мураками К., Юмото Ф, Оки С.Ю., Ясунага Т., Танокура М., Вакабаяси Т. Структурная основа Са2 + -регулируемой мышечной релаксации в местах взаимодействия тропонина с актином и тропомиозином. J Mol Biol 2005; 352 : 178–201.
CAS
Статья
Google Scholar
Такеда С., Ямасита А., Маеда К., Маеда Ю. Структура основного домена сердечного тропонина человека в Ca (2 +) — насыщенной форме. Nature 2003; 424 : 35–41.
Nature 2003; 424 : 35–41.
CAS
Статья
Google Scholar
Дистальный артрогрипоз с нарушением проприоцепции и осязания: описание раннего фенотипа у мальчика со сложной гетерозиготностью мутаций PIEZO2 и обзор литературы — FullText — Molecular Syndromology 2018, Vol.9, № 6
Аннотация
Рецессивное заболевание, ассоциированное с PIEZO2 , дистальный артрогрипоз с нарушением проприоцепции и осязания (DAIPT), характеризуется гипотонией, перинатальным респираторным дистрессом, значительно задержкой моторных вех и прогрессирующими симптомами дистального артрогрипоза и сколиоза. Здесь мы описываем самого молодого пациента с ДАИПТ на сегодняшний день, у которого в возрасте 3,5 лет не было ни одного клинического признака дистального артрогрипоза или контрактур, но в анамнезе были операции на двусторонней косолапости.Напротив, у него были некоторые признаки, не описанные до сих пор, такие как сирингогидромиелия, небольшая киста спинного мозга, умеренная микроцефалия с преждевременным закрытием переднего родничка и спонтанный односторонний вывих надколенника в возрасте 32 месяцев. Используя полное секвенирование экзома, мы идентифицировали 2 новые различные мутации потери функции в гене PIEZO2 у нашего пациента. Мы также рассматриваем фенотипы всех 16 ранее опубликованных пациентов с DAIPT, суммируем отличительные клинические особенности этого редкого генетического заболевания и рекомендуем включить DAIPT в дифференциальную диагностику гибкого младенца.PIEZO2 — это уникальный ионный канал, который преобразует механические импульсы в клеточные сигналы и участвует в различных путях механотрансдукции. В дополнение к DAIPT, мутации в PIEZO2 , как было описано, вызывают еще 3 различных фенотипа дистального артрогрипоза, которые являются доминантными и связаны с мутациями с усилением функции. Напротив, рецессивный DAIPT связан с мутациями потери функции PIEZO2 .
Используя полное секвенирование экзома, мы идентифицировали 2 новые различные мутации потери функции в гене PIEZO2 у нашего пациента. Мы также рассматриваем фенотипы всех 16 ранее опубликованных пациентов с DAIPT, суммируем отличительные клинические особенности этого редкого генетического заболевания и рекомендуем включить DAIPT в дифференциальную диагностику гибкого младенца.PIEZO2 — это уникальный ионный канал, который преобразует механические импульсы в клеточные сигналы и участвует в различных путях механотрансдукции. В дополнение к DAIPT, мутации в PIEZO2 , как было описано, вызывают еще 3 различных фенотипа дистального артрогрипоза, которые являются доминантными и связаны с мутациями с усилением функции. Напротив, рецессивный DAIPT связан с мутациями потери функции PIEZO2 .
© 2018 S. Karger AG, Базель
Дистальный артрогрипоз с нарушением проприоцепции и осязания (DAIPT; OMIM 617146) был впервые описан в 2016 г. [Delle Vedove et al., 2016] у 10 пациентов из 4 кровно-кровных семей, у которых был выявлен дистальный артрогрипоз, прогрессирующий сколиоз, значительно отсроченные двигательные этапы, тяжелая гипотония с мышечной атрофией и дизартрия наряду с нормальным интеллектуальным развитием и преходящими респираторными проблемами в раннем младенчестве. Все пациенты несли гомозиготные мутации потери функции в PIEZO2 (компонент 2 механочувствительного ионного канала пьезо-типа; OMIM 613629). Широкий спектр сенсорных и кинематических функций был изучен у 2 пациентов, у которых обоих были сложные гетерозиготные мутации в гене PIEZO2 [Chesler et al., 2016]. Махмуд и др. [2017] и Haliloglu et al. [2017] сообщили еще о 4 пациентах из 2 семей. Гетерозиготные носители (родители, братья и сестры) были здоровыми. Напротив, доминантные мутации с усилением функции PIEZO2 приводят к дистальному артрогрипозу 3 типа (DA3 или синдром Гордона с волчьей пастью, OMIM 114300), дистальному артрогрипозу 5 типа (DA5 с аномалиями глаза, OMIM 108145) или мардену.
[Delle Vedove et al., 2016] у 10 пациентов из 4 кровно-кровных семей, у которых был выявлен дистальный артрогрипоз, прогрессирующий сколиоз, значительно отсроченные двигательные этапы, тяжелая гипотония с мышечной атрофией и дизартрия наряду с нормальным интеллектуальным развитием и преходящими респираторными проблемами в раннем младенчестве. Все пациенты несли гомозиготные мутации потери функции в PIEZO2 (компонент 2 механочувствительного ионного канала пьезо-типа; OMIM 613629). Широкий спектр сенсорных и кинематических функций был изучен у 2 пациентов, у которых обоих были сложные гетерозиготные мутации в гене PIEZO2 [Chesler et al., 2016]. Махмуд и др. [2017] и Haliloglu et al. [2017] сообщили еще о 4 пациентах из 2 семей. Гетерозиготные носители (родители, братья и сестры) были здоровыми. Напротив, доминантные мутации с усилением функции PIEZO2 приводят к дистальному артрогрипозу 3 типа (DA3 или синдром Гордона с волчьей пастью, OMIM 114300), дистальному артрогрипозу 5 типа (DA5 с аномалиями глаза, OMIM 108145) или мардену. -Синдром Уокера (OMIM 248700) [Coste et al., 2013; McMillin et al., 2014]. Здесь мы сообщаем о самом молодом пациенте с DAIPT, который наблюдался на сегодняшний день, выявляя отчетливый ранний фенотип с некоторыми новыми особенностями, и анализируем клинические характеристики пациентов с DAIPT из литературы.
-Синдром Уокера (OMIM 248700) [Coste et al., 2013; McMillin et al., 2014]. Здесь мы сообщаем о самом молодом пациенте с DAIPT, который наблюдался на сегодняшний день, выявляя отчетливый ранний фенотип с некоторыми новыми особенностями, и анализируем клинические характеристики пациентов с DAIPT из литературы.
Клинический отчет
Мужчина европеоидной расы 3,5 лет (2013 г.р.) был направлен в наш Институт медицинской генетики из Центра возрастной неврологии и социальной педиатрии для генетической оценки и диагностики из-за гипотонии со значительно задержанными двигательными этапами.
Беременность протекала без осложнений до тех пор, пока при ультразвуковом обследовании органов (20 неделя беременности) не было выявлено двустороннее эквиноварусное сочленение стопы. На МРТ плода других дополнительных патологий не выявлено.Мать отметила, что шевеление плода могло быть уменьшено. Вскоре после рождения (40-я неделя беременности, 3566 г, 52 см, OFC 33 см, оценка по шкале Апгар 9/10/10) присутствовали стридор и одышка, а также тяжелая гипотония, слабость и цианоз во время кормления. Новорожденный в возрасте 1 недели был описан как «имеющий колоколообразную грудную клетку, почти без спонтанного дыхания (требовалась энергичная стимуляция), с очень редкими спонтанными движениями даже после болезненного импульса». Мальчику требовалась интенсивная терапия и кислородная поддержка в течение 2 месяцев, дважды переболел пневмонией.Бронхоскопия не выявила каких-либо структурных аномалий в дыхательных путях, хотя в конце вдоха наблюдалось почти полное закрытие дыхательных путей в областях гипофаринкса и aditus laryngis. В течение первых 2 месяцев было назначено кормление через назогастральный зонд, и у мальчика была сильная диарея и рефлюкс, которые позже улучшились. Ранние ультразвуковые исследования головного мозга, сердца и брюшной полости были нормальными, как и проверка слуха, офтальмологическое обследование, оценка метаболизма и массив-анализ CGH.Два МРТ головного мозга и позвоночника были выполнены в разное время (МРТ головного мозга в возрасте 1 месяц при госпитализации в отделении интенсивной терапии новорожденных и в возрасте 1 год и 6 месяцев в рамках неврологического обследования; позвоночник МРТ через 1 год / 2 месяца и 1 год / 6 месяцев).
Новорожденный в возрасте 1 недели был описан как «имеющий колоколообразную грудную клетку, почти без спонтанного дыхания (требовалась энергичная стимуляция), с очень редкими спонтанными движениями даже после болезненного импульса». Мальчику требовалась интенсивная терапия и кислородная поддержка в течение 2 месяцев, дважды переболел пневмонией.Бронхоскопия не выявила каких-либо структурных аномалий в дыхательных путях, хотя в конце вдоха наблюдалось почти полное закрытие дыхательных путей в областях гипофаринкса и aditus laryngis. В течение первых 2 месяцев было назначено кормление через назогастральный зонд, и у мальчика была сильная диарея и рефлюкс, которые позже улучшились. Ранние ультразвуковые исследования головного мозга, сердца и брюшной полости были нормальными, как и проверка слуха, офтальмологическое обследование, оценка метаболизма и массив-анализ CGH.Два МРТ головного мозга и позвоночника были выполнены в разное время (МРТ головного мозга в возрасте 1 месяц при госпитализации в отделении интенсивной терапии новорожденных и в возрасте 1 год и 6 месяцев в рамках неврологического обследования; позвоночник МРТ через 1 год / 2 месяца и 1 год / 6 месяцев). В то время как никаких аномалий в головном мозге не наблюдалось (микроцефалия не упоминалась, вероятно, из-за незначительного отклонения), в спинном мозге наблюдалась умеренная эктазия центрального канала в виде сирингогидромиелии и небольшое кистозное поражение в нижней части шейного отдела позвоночника. похож на нейрентерическую или арахноидальную кисту, без признаков прогрессирования между визуализацией.Неврологическое обследование показало гипорефлексию (11 месяцев), а затем арефлексию (2 года) нижних конечностей без пирамидных признаков (Бабинский, Оппенгейм), что не соответствует диагнозу нервно-мышечного заболевания или нарушения 2-го мотонейрона. Судорожных припадков у мальчика не было, на ЭЭГ патологий не выявлено. Его социальное развитие и зрительный контакт были очень хорошими, хотя основные двигательные вехи были значительно отложены из-за тяжелой гипотонии; пациент мог переворачиваться примерно в 10 месяцев, сидеть в 19 месяцев и ползать в 2 года / 3 месяца, но он не мог ходить.
В то время как никаких аномалий в головном мозге не наблюдалось (микроцефалия не упоминалась, вероятно, из-за незначительного отклонения), в спинном мозге наблюдалась умеренная эктазия центрального канала в виде сирингогидромиелии и небольшое кистозное поражение в нижней части шейного отдела позвоночника. похож на нейрентерическую или арахноидальную кисту, без признаков прогрессирования между визуализацией.Неврологическое обследование показало гипорефлексию (11 месяцев), а затем арефлексию (2 года) нижних конечностей без пирамидных признаков (Бабинский, Оппенгейм), что не соответствует диагнозу нервно-мышечного заболевания или нарушения 2-го мотонейрона. Судорожных припадков у мальчика не было, на ЭЭГ патологий не выявлено. Его социальное развитие и зрительный контакт были очень хорошими, хотя основные двигательные вехи были значительно отложены из-за тяжелой гипотонии; пациент мог переворачиваться примерно в 10 месяцев, сидеть в 19 месяцев и ползать в 2 года / 3 месяца, но он не мог ходить. Его мелкая моторика развивалась намного лучше, а его речь и интеллектуальное развитие соответствовали возрасту. Пациент находился в 50-м процентиле (P) по длине и весу; однако OFC был немного ниже P3, а передний родничок закрылся преждевременно (4 месяца). Операция по поводу эквиноварусной стопы была произведена в возрасте 6 месяцев на обеих стопах после гипсовой терапии. Пациенту также была сделана односторонняя орхидопексия. Правовыпуклый сколиоз с вершиной в грудопоясничном переходе был диагностирован в возрасте 2 лет, пациентка лечилась спинным корсетом.В возрасте 2 лет / 8 месяцев мальчик проснулся однажды утром с болезненной опухолью левого колена без какой-либо известной травмы. После первоначального подозрения на ювенильный артрит был диагностирован спонтанный вывих надколенника, который позже потребовал хирургической коррекции.
Его мелкая моторика развивалась намного лучше, а его речь и интеллектуальное развитие соответствовали возрасту. Пациент находился в 50-м процентиле (P) по длине и весу; однако OFC был немного ниже P3, а передний родничок закрылся преждевременно (4 месяца). Операция по поводу эквиноварусной стопы была произведена в возрасте 6 месяцев на обеих стопах после гипсовой терапии. Пациенту также была сделана односторонняя орхидопексия. Правовыпуклый сколиоз с вершиной в грудопоясничном переходе был диагностирован в возрасте 2 лет, пациентка лечилась спинным корсетом.В возрасте 2 лет / 8 месяцев мальчик проснулся однажды утром с болезненной опухолью левого колена без какой-либо известной травмы. После первоначального подозрения на ювенильный артрит был диагностирован спонтанный вывих надколенника, который позже потребовал хирургической коррекции.
На сегодняшний день у пациента нет братьев и сестер, а его родители — здоровые, не кровные кавказцы. Физикальное обследование в возрасте 3,5 лет показало пограничный низкий уровень OFC (рост 97,5 см / P30, вес 14 кг / P24 и OFC 47 см / чуть ниже P3). Пациент мог сидеть, ползать и играть на земле, но не мог стоять самостоятельно (рис. 1а). Разговорный язык был логичным и состоял из полных предложений, хотя его речь была гнусавой, невнятной и иногда трудной для понимания. У него была обнаружена умеренная грудная клетка (рис. 1b, d), сколиоз, alatae лопатки и значительная глобальная гипотония. Суставы были умеренно гипермобильными, левое колено опухшее с латерально смещенной надколенником (рис. 1а, б). В остальном конечности были симметричными, без контрактур или каких-либо других патологий, за исключением легкой степени плоскостопия (рис.1e-h). Его кожный сосудистый рисунок в верхней части груди и лица был более выраженным, тогда как избыточные кожные складки наблюдались на груди и животе (рис. 1c, d). Соски были широко расставленными, маленькими и антивёрнутыми. Грыжи живота отсутствовали; печень и селезенка не пальпировались, гениталии соответствовали возрасту. Наблюдались некоторые тонкие дисморфические черепно-лицевые особенности (рис.
Пациент мог сидеть, ползать и играть на земле, но не мог стоять самостоятельно (рис. 1а). Разговорный язык был логичным и состоял из полных предложений, хотя его речь была гнусавой, невнятной и иногда трудной для понимания. У него была обнаружена умеренная грудная клетка (рис. 1b, d), сколиоз, alatae лопатки и значительная глобальная гипотония. Суставы были умеренно гипермобильными, левое колено опухшее с латерально смещенной надколенником (рис. 1а, б). В остальном конечности были симметричными, без контрактур или каких-либо других патологий, за исключением легкой степени плоскостопия (рис.1e-h). Его кожный сосудистый рисунок в верхней части груди и лица был более выраженным, тогда как избыточные кожные складки наблюдались на груди и животе (рис. 1c, d). Соски были широко расставленными, маленькими и антивёрнутыми. Грыжи живота отсутствовали; печень и селезенка не пальпировались, гениталии соответствовали возрасту. Наблюдались некоторые тонкие дисморфические черепно-лицевые особенности (рис. 1b-d), включая плоский и асимметричный затылок и широкий лоб, в отличие от заостренного и несколько асимметричного подбородка.Уши были на границе низко посажены, но хорошо сформированы, а глаза были посажены немного глубже, имели миндалевидную форму и имели короткие глазные щели и эпикантус. Желобок был длинным, а нёбо высоким. Зубы и шея без патологий. Во время последней консультации родители отметили, что мальчик (4 года / 3 месяца) все еще не может ходить.
1b-d), включая плоский и асимметричный затылок и широкий лоб, в отличие от заостренного и несколько асимметричного подбородка.Уши были на границе низко посажены, но хорошо сформированы, а глаза были посажены немного глубже, имели миндалевидную форму и имели короткие глазные щели и эпикантус. Желобок был длинным, а нёбо высоким. Зубы и шея без патологий. Во время последней консультации родители отметили, что мальчик (4 года / 3 месяца) все еще не может ходить.
Рис. 1
Фенотип больного (возраст 3,5 года). a , b Все тело и конечности.Показаны гипотония, неспособность самостоятельно стоять или ходить и вывих левой надколенника. c , d Лицо и верхняя часть тела. Заметны слегка короткие глазные щели, заостренный асимметричный подбородок, умеренная грудная клетка, втянутые соски и выраженный сосудистый рисунок в верхней части грудной клетки, а также дряблая кожа в нижней части груди. e-h Руки и ступни без контрактур. Произведена хирургическая коррекция двусторонней косолапости в возрасте 6 месяцев.
Материалы и методы
Полную библиотеку секвенирования экзома получали с помощью набора TruSeq Exome (Illumina, Сан-Диего, Калифорния, США) и далее анализировали с использованием системы секвенирования NextSeq 500 (Illumina).Биоинформатическую оценку выполняли с использованием программного обеспечения VarSeq (Golden Helix, Bozeman, MT, USA), и 94% проанализированных последовательностей были покрыты> 20 раз. Различные базы данных (ExAC, EVS, 1000 Genomes, HGMD и OMIM) использовались для оценки частотности вариантов и / или их влияния. Варианты были отфильтрованы в соответствии с их возможной ассоциацией с фенотипом пациента, и при необходимости их патогенность оценивалась с помощью 6 программ прогнозирования (SIFT, Polyphen2, MutationTaster, MutationAssessor, FATHMM и FATHMM MKL Coding).Все соответствующие мутации были подтверждены секвенированием по Сэнгеру.
Результаты
Посредством анализа секвенирования всего экзома были идентифицированы 2 сложные гетерозиготные мутации в гене PIEZO2 пациента, обе из которых, как было предсказано, вызывают потерю функции на уровне белка (рис. 2a, b). Первая мутация, c.76C> T (NM_022068.3), расположена в экзоне 2 и, по прогнозам, продуцирует преждевременный стоп-кодон, p.Arg26Ter (NP_071351.2). Он не был указан в ExAC и имел частоту 0.0002 в 1000 геномах. Вторая мутация, c.1528-1G> T, локализуется в высококонсервативном динуклеотиде акцепторного сайта сплайсинга в интроне 12 и, как предполагается, влияет на сплайсинг. Он не был указан в ExAC или 1000 Genomes. Секвенирование по Сэнгеру показало, что каждый родитель несет 1 гетерозиготную мутацию (c.76C> T у матери и c.1528-1G> T у отца). Наиболее важные фенотипические особенности DAIPT, основанные на описанных 16 пациентах из 8 семей и нашем пациенте, перечислены в таблицах 1, 2.
2a, b). Первая мутация, c.76C> T (NM_022068.3), расположена в экзоне 2 и, по прогнозам, продуцирует преждевременный стоп-кодон, p.Arg26Ter (NP_071351.2). Он не был указан в ExAC и имел частоту 0.0002 в 1000 геномах. Вторая мутация, c.1528-1G> T, локализуется в высококонсервативном динуклеотиде акцепторного сайта сплайсинга в интроне 12 и, как предполагается, влияет на сплайсинг. Он не был указан в ExAC или 1000 Genomes. Секвенирование по Сэнгеру показало, что каждый родитель несет 1 гетерозиготную мутацию (c.76C> T у матери и c.1528-1G> T у отца). Наиболее важные фенотипические особенности DAIPT, основанные на описанных 16 пациентах из 8 семей и нашем пациенте, перечислены в таблицах 1, 2.
Таблица 1
Клинические особенности DAIPT
Таблица 2
Опубликованные пациенты с DAIPT и гомозиготными / сложными гетерозиготными мутациями в PIEZO2
Рис. 2
Последовательности мутаций 09 PIEZO пациента. a Мутация c.76C> T (p.Arg26Ter) в экзоне 2. b Мутация c.1528-1G> T в интроне 12.
b Мутация c.1528-1G> T в интроне 12.
Обсуждение
Ген PIEZO2 расположен в хромосоме 18 (18p11 .22), его размер 477 т.п.н., он включает 52 экзона. Размер транскрипта основного гена (ENST00000503781.7) составляет 8 259 п.н., а кодируемый белок состоит из 2 752 аминокислот. Мутации в PIEZO2 могут относиться либо к типу увеличения функции, вызывающему доминантные фенотипы дистального артрогрипоза, либо по типу потери функции, вызывающему рецессивный DAIPT. Большинство доминантных мутаций PIEZO2 сгруппированы в 2 регионах гена, которые кодируют С-концевой домен и, по-видимому, являются горячими точками мутаций, особенно в последнем экзоне 52.Напротив, мутации в рецессивном DAIPT не обнаруживают какой-либо локусной предрасположенности, поскольку они распределены по всему гену [Chesler et al., 2016; Delle Vedove et al., 2016; Haliloglu et al., 2017; Махмуд и др., 2017]. Белок PIEZO2 содержит более 30 трансмембранных доменов и функционирует как часть механически активируемых катионных каналов [Coste et al.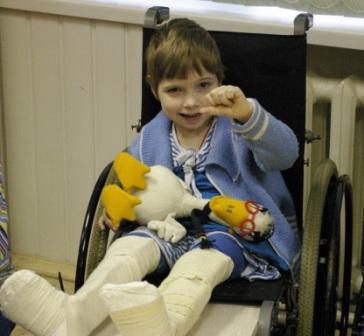 , 2010]. Единственный другой известный белок с аналогичной функцией механотрансдукции у людей, PIEZO1, кодируется геном PIEZO1 , , который имеет примерно 50% своей последовательности ДНК с PIEZO2.PIEZO1 также обнаруживает доминантные мутации с усилением функции и рецессивные мутации с потерей функции, которые вызывают различные фенотипы, а именно дегидратированный наследственный стоматоцитоз с гемолитической анемией (доминантная) и генерализованная лимфатическая дисплазия (рецессивная), но не артрогрипоз [Zarychanski et al. , 2012; Ли и др., 2014; Фотиу и др., 2015]. PIEZO2 и PIEZO1 — очень большие, сложные и консервативные трансмембранные белки [Coste et al., 2010]. Их также называют каналами, активируемыми растяжением, и они присутствуют в различных типах клеток, в которых механические стимулы необходимо преобразовывать в электрические сигналы, например, в соматосенсорных нейронах, мышечных клетках, остеобластах, эндотелиальных клетках и сенсорных клетках внутреннего уха [Coste и другие.
, 2010]. Единственный другой известный белок с аналогичной функцией механотрансдукции у людей, PIEZO1, кодируется геном PIEZO1 , , который имеет примерно 50% своей последовательности ДНК с PIEZO2.PIEZO1 также обнаруживает доминантные мутации с усилением функции и рецессивные мутации с потерей функции, которые вызывают различные фенотипы, а именно дегидратированный наследственный стоматоцитоз с гемолитической анемией (доминантная) и генерализованная лимфатическая дисплазия (рецессивная), но не артрогрипоз [Zarychanski et al. , 2012; Ли и др., 2014; Фотиу и др., 2015]. PIEZO2 и PIEZO1 — очень большие, сложные и консервативные трансмембранные белки [Coste et al., 2010]. Их также называют каналами, активируемыми растяжением, и они присутствуют в различных типах клеток, в которых механические стимулы необходимо преобразовывать в электрические сигналы, например, в соматосенсорных нейронах, мышечных клетках, остеобластах, эндотелиальных клетках и сенсорных клетках внутреннего уха [Coste и другие. , 2010; Сакс, 2010; Пардо-Пастор и др., 2018]. У человека PIEZO2 является основным механотрансдуктором проприорецепторов [Woo et al., 2015]. Проприоцепция, восприятие положения тела и конечностей, опосредуется специализированными механосенсорными нейронами, которые передают информацию о растяжении и напряжении мышц, сухожилий, кожи и суставов. Мутации потери функции и увеличения функции согласуются с симптоматикой опорно-двигательного аппарата при заболеваниях, связанных с PIEZO2 , при которых как пониженная, так и повышенная активность белка PIEZO2 имеют различные отрицательные эффекты.PIEZO2 также является универсальным датчиком растяжения дыхательных путей при раздувании легких, и он имеет решающее значение для правильного расширения легких, обеспечения эффективного дыхания при рождении и поддержания нормального дыхания у взрослых мышей [Nonomura et al., 2017]. Эти данные совместимы с человеческими фенотипами, ассоциированными с PIEZO2 , в отношении респираторного дистресса в неонатальном периоде у пациентов с DAIPT, а также в отношении прогрессирующего рестриктивного заболевания легких у взрослых с DA5.
, 2010; Сакс, 2010; Пардо-Пастор и др., 2018]. У человека PIEZO2 является основным механотрансдуктором проприорецепторов [Woo et al., 2015]. Проприоцепция, восприятие положения тела и конечностей, опосредуется специализированными механосенсорными нейронами, которые передают информацию о растяжении и напряжении мышц, сухожилий, кожи и суставов. Мутации потери функции и увеличения функции согласуются с симптоматикой опорно-двигательного аппарата при заболеваниях, связанных с PIEZO2 , при которых как пониженная, так и повышенная активность белка PIEZO2 имеют различные отрицательные эффекты.PIEZO2 также является универсальным датчиком растяжения дыхательных путей при раздувании легких, и он имеет решающее значение для правильного расширения легких, обеспечения эффективного дыхания при рождении и поддержания нормального дыхания у взрослых мышей [Nonomura et al., 2017]. Эти данные совместимы с человеческими фенотипами, ассоциированными с PIEZO2 , в отношении респираторного дистресса в неонатальном периоде у пациентов с DAIPT, а также в отношении прогрессирующего рестриктивного заболевания легких у взрослых с DA5.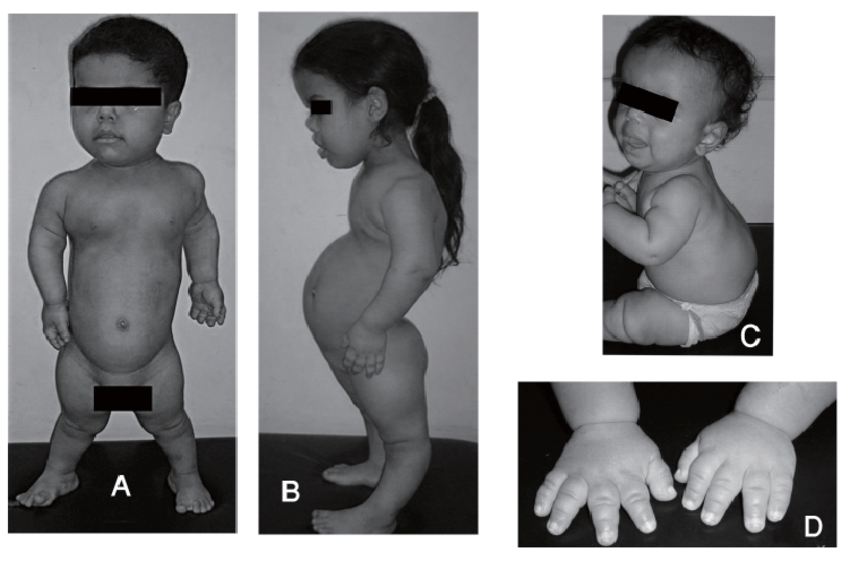
Мутации с двуаллельной потерей функции в PIEZO2 вызывают специфический фенотип DAIPT, включая тяжелую гипотонию со значительной задержкой двигательных этапов, временный респираторный дистресс и проблемы с кормлением в раннем младенчестве, а также симптомы тяжелого прогрессирующего сколиоза и прогрессирующие деформации контрактуры кистей и стоп (дистальный артрогрипоз).Сравнивая признаки рецессивных и доминантных заболеваний, ассоциированных с PIEZO2- , дистальный артрогрипоз при доминантных формах обычно является врожденным, тогда как сколиоз встречается реже, только у 16 из 61 пациента [McMillin et al., 2014]. Проблемы с дыханием и кормлением в младенчестве еще не описаны в случае доминирующих заболеваний, связанных с PIEZO2-, но прогрессирующее рестриктивное заболевание легких у взрослых наблюдалось в DA5. Интеллектуальное развитие является нормальным также при доминирующих заболеваниях, связанных с PIEZO2- , за исключением наиболее тяжелой и редкой формы, синдрома Мардена-Уокера, который помимо контрактур суставов и кифосколиоза включает множество дополнительных аномалий (маскоподобное лицо, блефарофимоз, микрогнатия, волчья пасть, деформации грудной клетки, нарушение нормального роста, почечные и церебральные аномалии). Однако на сегодняшний день подтверждено, что только один пациент с синдромом Мардена-Уокера несет доминантную мутацию PIEZO2 [McMillin et al., 2014]. DA3 (синдром Гордона) включает дополнительную волчью нёбо или раздвоенный язычок. У пациентов с DA5 также проявляются различные глазные симптомы (например, офтальмоплегия, птоз, глубоко посаженные глаза, блефарофимоз и аномалии сетчатки) и прогрессирующее рестриктивное заболевание легких [McMillin et al., 2014; Alisch et al., 2017]. Однако некоторые особенности доминирующих заболеваний, ассоциированных с PIEZO2 , частично совпадают.Некоторые аномалии мозга, описанные при синдроме Мардена-Уокера (мальформация Денди-Уокера или Арнольда-Киари), также случайно были отмечены как в DA3, так и в DA5 [McMillin et al., 2014].
Однако на сегодняшний день подтверждено, что только один пациент с синдромом Мардена-Уокера несет доминантную мутацию PIEZO2 [McMillin et al., 2014]. DA3 (синдром Гордона) включает дополнительную волчью нёбо или раздвоенный язычок. У пациентов с DA5 также проявляются различные глазные симптомы (например, офтальмоплегия, птоз, глубоко посаженные глаза, блефарофимоз и аномалии сетчатки) и прогрессирующее рестриктивное заболевание легких [McMillin et al., 2014; Alisch et al., 2017]. Однако некоторые особенности доминирующих заболеваний, ассоциированных с PIEZO2 , частично совпадают.Некоторые аномалии мозга, описанные при синдроме Мардена-Уокера (мальформация Денди-Уокера или Арнольда-Киари), также случайно были отмечены как в DA3, так и в DA5 [McMillin et al., 2014].
Следующая спецификация фенотипа DAIPT основана на 16 опубликованных на данный момент пациентах плюс наш пациент и включает тяжелую гипотонию с двигательной задержкой и нормальным интеллектом, тяжелый сколиоз, прогрессирующий дистальный артрогрипоз и некоторые другие особенности (Таблица 1, 2).
 Большинство этих генов кодируют структурные компоненты сократительного аппарата быстросокращающихся миофибрилл: MYh4 кодирует эмбриональный миозин, TNNI2 тропонин I, TNNT3 тропонин Т, TPM2 тропомиозин 2 тяжелой цепи и периносатальный тяжелый скелет 8 цепи MYH8 ) [Toydemir et al., 2006а, б; Sung et al., 2003a, b]. Ген MYBPC1 кодирует миозин-связывающий протеин C медленного типа; ECEL1 кодирует одну из металлопротеаз цинка, экспрессируемых во время эмбриогенеза в ЦНС, а также в скелетных мышцах, а FBN2 кодирует фибриллин-2, часть микрофибрилл эластических волокон, важных для раннего эластогенеза в коже, связках, мышцах и др. ткани [Putnam et al., 1995; Gurnett et al., 2010; Dieterich et al., 2013]. Ранний и прогрессирующий сколиоз (начинающийся в возрасте от 1 до 5 лет) был серьезной проблемой для здоровья, отмеченной у всех пациентов с DAIPT, иногда даже требующих хирургического вмешательства.Почти 80% пациентов (11/14) имели низкий рост.
Большинство этих генов кодируют структурные компоненты сократительного аппарата быстросокращающихся миофибрилл: MYh4 кодирует эмбриональный миозин, TNNI2 тропонин I, TNNT3 тропонин Т, TPM2 тропомиозин 2 тяжелой цепи и периносатальный тяжелый скелет 8 цепи MYH8 ) [Toydemir et al., 2006а, б; Sung et al., 2003a, b]. Ген MYBPC1 кодирует миозин-связывающий протеин C медленного типа; ECEL1 кодирует одну из металлопротеаз цинка, экспрессируемых во время эмбриогенеза в ЦНС, а также в скелетных мышцах, а FBN2 кодирует фибриллин-2, часть микрофибрилл эластических волокон, важных для раннего эластогенеза в коже, связках, мышцах и др. ткани [Putnam et al., 1995; Gurnett et al., 2010; Dieterich et al., 2013]. Ранний и прогрессирующий сколиоз (начинающийся в возрасте от 1 до 5 лет) был серьезной проблемой для здоровья, отмеченной у всех пациентов с DAIPT, иногда даже требующих хирургического вмешательства.Почти 80% пациентов (11/14) имели низкий рост. Более чем у половины пациентов (9/16) наблюдалась арахнодактилия. Большинство пациентов DAIPT (11/15) приобрели способность ходить, хотя и значительно позже (от 5 до 16 лет). Описана неуверенная походка на широкую ногу, возможная только с открытыми глазами. У всех пациентов отсутствовали глубокие сухожильные рефлексы. МРТ головного и спинного мозга были нормальными, только у нашего пациента была легкая сирингогидромиелия и небольшое кистозное поражение в нижнем отделе позвоночника без прогрессирования.Все пациенты, прошедшие тестирование скорости нервной проводимости или проприоцепции (5/5 и 7/7 соответственно), показали легкую сенсорную полинейропатию и / или нарушение проприоцепции. У некоторых людей (3/6) временно умеренно повышенная креатинкиназа. Не было особых офтальмологических проблем и расщелины неба по сравнению с пациентами с доминантным дистальным артрогрипозом из-за мутации PIEZO2 . Однако у некоторых пациентов с DAIPT было высокое небо и легкий птоз или гипомимия.
Более чем у половины пациентов (9/16) наблюдалась арахнодактилия. Большинство пациентов DAIPT (11/15) приобрели способность ходить, хотя и значительно позже (от 5 до 16 лет). Описана неуверенная походка на широкую ногу, возможная только с открытыми глазами. У всех пациентов отсутствовали глубокие сухожильные рефлексы. МРТ головного и спинного мозга были нормальными, только у нашего пациента была легкая сирингогидромиелия и небольшое кистозное поражение в нижнем отделе позвоночника без прогрессирования.Все пациенты, прошедшие тестирование скорости нервной проводимости или проприоцепции (5/5 и 7/7 соответственно), показали легкую сенсорную полинейропатию и / или нарушение проприоцепции. У некоторых людей (3/6) временно умеренно повышенная креатинкиназа. Не было особых офтальмологических проблем и расщелины неба по сравнению с пациентами с доминантным дистальным артрогрипозом из-за мутации PIEZO2 . Однако у некоторых пациентов с DAIPT было высокое небо и легкий птоз или гипомимия. Когнитивное развитие обычно было нормальным.Только в одной семье умеренная когнитивная инвалидность была описана у 2 затронутых детей, на что, возможно, также повлиял низкий социально-экономический статус семьи [Delle Vedove et al., 2016]. Гетерозиготные носители мутаций потери функции PIEZO2 (родители, братья и сестры) были здоровыми, без каких-либо признаков нервно-мышечной или скелетной симптоматики.
Когнитивное развитие обычно было нормальным.Только в одной семье умеренная когнитивная инвалидность была описана у 2 затронутых детей, на что, возможно, также повлиял низкий социально-экономический статус семьи [Delle Vedove et al., 2016]. Гетерозиготные носители мутаций потери функции PIEZO2 (родители, братья и сестры) были здоровыми, без каких-либо признаков нервно-мышечной или скелетной симптоматики.
У нашего пациента мы наблюдали несколько особенностей, которые еще не были описаны в DAIPT, такие как пограничная микроцефалия с преждевременным закрытием переднего родничка и асимметрия головы, легкая сирингидромиелия с небольшой кистой, спонтанный вывих левой надколенника в возрасте 2 года 8 месяцев, без травм и крипторхизма.Ни у одного другого пациента не было выявлено аномалий спинного мозга с помощью МРТ, и оценка того, связана ли сирингомиелия, наблюдаемая у нашего пациента, с мутациями PIEZO2 , в настоящее время невозможна.
В заключение, мы предполагаем, что в раннем возрасте фенотип DAIPT может быть довольно неспецифичным, поскольку он характеризуется клинической картиной вялого младенца с респираторным дистресс-синдромом и двусторонними деформациями стопы. Эти особенности могут проявляться в различных моногенных нарушениях (например, немалиновых миопатиях, последовательности акинезии плода, летальных врожденных контрактурах, дистальной спинномозговой мышечной атрофии 1 и др.).Типичные признаки контрактур кисти и пальцев при DAIPT, вероятно, развиваются в более позднем возрасте, как и сколиоз. Моторное и когнитивное развитие также невозможно оценить в раннем младенчестве. Таким образом, вполне возможно, что большинство очень молодых пациентов с ДАИПТ останутся недиагностированными, что может помешать раннему лечению сколиоза, а также уведомлению семьи о 25% риске рецидива. Мы рекомендуем включить DAIPT в дифференциальную диагностику гибкого ребенка, особенно при наличии дополнительных деформаций стопы и респираторного дистресс-синдрома.Учитывая отсутствие клинической специфичности для DAIPT в молодом возрасте и большой размер гена PIEZO2 , полное секвенирование экзома после исключения наиболее распространенных генетических причин тяжелой неонатальной гипотонии (например, хромосомных аномалий, спинальной мышечной атрофии, Прадера- Синдром Вилли и миотоническая дистрофия 1 типа) — наиболее эффективный метод диагностики ДАИПТ в раннем детстве.
Эти особенности могут проявляться в различных моногенных нарушениях (например, немалиновых миопатиях, последовательности акинезии плода, летальных врожденных контрактурах, дистальной спинномозговой мышечной атрофии 1 и др.).Типичные признаки контрактур кисти и пальцев при DAIPT, вероятно, развиваются в более позднем возрасте, как и сколиоз. Моторное и когнитивное развитие также невозможно оценить в раннем младенчестве. Таким образом, вполне возможно, что большинство очень молодых пациентов с ДАИПТ останутся недиагностированными, что может помешать раннему лечению сколиоза, а также уведомлению семьи о 25% риске рецидива. Мы рекомендуем включить DAIPT в дифференциальную диагностику гибкого ребенка, особенно при наличии дополнительных деформаций стопы и респираторного дистресс-синдрома.Учитывая отсутствие клинической специфичности для DAIPT в молодом возрасте и большой размер гена PIEZO2 , полное секвенирование экзома после исключения наиболее распространенных генетических причин тяжелой неонатальной гипотонии (например, хромосомных аномалий, спинальной мышечной атрофии, Прадера- Синдром Вилли и миотоническая дистрофия 1 типа) — наиболее эффективный метод диагностики ДАИПТ в раннем детстве.
Благодарности
Мы благодарим семью за готовность принять участие в этом отчете.Мы также благодарим Марион Хагл за техническую помощь.
Заявление об этике
Информированное согласие было получено до проведения генетического анализа и публикации клинических деталей этого исследования. Авторы не раскрывают никаких этических конфликтов.
Заявление о раскрытии информации
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
- Alisch F, Weichert A, Kalche K, Paradiso V, Longardt AC и др.: Семейный синдром Гордона, связанный с мутацией PIEZO2 .Am J Med Genet A 173: 254-259 (2017).
- Chesler AT, Szczot M, Bharucha-Goebel D, Čeko M, Donkervoort S, et al: Роль PIEZO2 в механочувствительности человека. N Engl J Med 375: 1355-1364 (2016).
- Косте Б., Матур Дж., Шмидт М., Эрли Т.Дж., Ранад С. и др.: Пьезо1 и Пьезо2 являются важными компонентами отдельных механически активируемых катионных каналов.Наука 330: 55-60 (2010).
- Косте Б., Хоуг Дж., Мюррей М.Ф., Стициэль Н., Банделл М. и др.: Мутации с усилением функции в механически активированном ионном канале PIEZO2 вызывают подтип дистального артрогрипоза. Proc Natl Acad Sci USA 110: 4667-4672 (2013).
- Делле Ведове А., Сторбек М., Хеллер Р., Хёлькер И., Хеббар М. и др.: Двуаллельная потеря связанного с проприоцепцией PIEZO2 вызывает мышечную атрофию с перинатальным респираторным дистрессом, артрогрипозом и сколиозом.Am J Hum Genet 99: 1206-1216 (2016).
- Дитрих К., Кихано-Рой С., Монье Н., Чжоу Дж., Форе Дж и др.: Эндопептидаза нейронов ECEL1 связана с особой формой рецессивного дистального артрогрипоза. Hum Mol Genet 22: 1483-1492 (2013).
- Фотиу Э, Мартин-Альмедина С., Симпсон М.А., Лин С., Гордон К. и др.: Новые мутации в PIEZO1 вызывают аутосомно-рецессивную генерализованную лимфатическую дисплазию с неиммунной водянкой плода.Нац Коммуна 6: 8085 (2015).
- Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI и др.: Миозин-связывающий белок C1: новый ген для аутосомно-доминантного дистального артрогрипоза типа 1. Hum Mol Genet 19: 1165-1173 (2010).
- Haliloglu G, Becker K, Temucin C, Talim B, Küçükşahin N, et al: Recessive PIEZO2 stop мутация вызывает дистальный артрогрипоз со слабостью дистальных мышц, сколиозом и дефектами проприоцепции.J Hum Genet 62: 497-501 (2017).
- Ли Дж., Хоу Б., Тумова С., Мураки К., Брунс А. и др.: Интеграция Piezo1 в сосудистую архитектуру с физиологической силой. Nature 515: 279-282 (2014).
- Махмуд А.А., Нахид Н.А., Нассиф К., Сайид М.С., Ахмед М.Ю. и др.: Потеря проприоцепции и канала сенсорных ощущений PIEZO2 у братьев и сестер с прогрессирующей формой контрактур.Clin Genet 91: 470-475 (2017).
- McMillin MJ, Beck AE, Chong JX, Shively KM, Buckingham KJ, et al: Мутации в PIEZO2 вызывают синдром Гордона, синдром Мардена-Уокера и дистальный тип артрогрипоза 5. Am J Hum Genet 94: 734-744 (2014) .
- Nonomura K, Woo SH, Chang RB, Gillich A, Qiu Z, et al: Piezo2 определяет растяжение дыхательных путей и опосредует апноэ, вызванное раздуванием легких.Nature 541: 176-181 (2017).
- Пардо-Пастор С., Рубио-Москардо Ф., Фогель-Гонсалес М., Серра С.А., Афтинос А. и др.: Канал Piezo2 регулирует цитоскелет RhoA и актина, способствуя механобиологическим ответам клеток. Proc Natl Acad Sci USA 115: 1925-1930 (2018).
- Putnam EA, Zhang H, Ramirez F, Milewicz DM: Мутации фибриллина-2 ( FBN2 ) приводят к марфановскому расстройству, врожденной контрактурной арахнодактилии.Нат Генет 11: 456-458 (1995).
- Сакс Ф .: Ионные каналы, активируемые растяжением: что это такое? Физиология (Bethesda) 25: 50-56 (2010).
- Sung SS, Brassington AME, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA и др.: Мутации в генах, кодирующих быстро сокращающиеся сократительные белки, вызывают синдромы дистального артрогрипоза.Am J Hum Genet 72: 681-690 (2003a).
- Sung SS, Brassington AM, Krakowiak PA, Carey JC, Jorde LB, Bamshad M: Мутации в TNNT3 вызывают множественные врожденные контрактуры: второй локус для дистального артрогрипоза типа 2B. Am J Hum Genet 73: 212-214 (2003b).
- Toydemir RM, Резерфорд A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ: Мутации в тяжелой цепи эмбрионального миозина ( MYh4 ) вызывают синдром Фримена-Шелдона и синдром Шелдона-Холла.Нат Генет 38: 561-565 (2006a).
- Toydemir RM, Chen H, Proud VK, Martin R, van Bokhoven H, et al: Синдром тризм-псевдокамптодактилии вызван повторяющейся мутацией MYH8 . Am J Med Genet 140: 2387-2393 (2006b).
- Woo SH, Lukacs V, de Nooij JC, Zaytseva D, Criddle CR, et al: Piezo2 является основным каналом механотрансдукции для проприоцепции.Nat Neurosci 18: 1756-1762 (2015).
- Зарычанский Р., Шульц В.П., Хьюстон Б.Л., Максимова Ю., Хьюстон Д.С. и др.: Мутации в белке механотрансдукции PIEZO1 связаны с наследственным ксероцитозом. Кровь 120: 1908-1915 (2012).
Автор Контакты
Яна Бехунова
Институт медицинской генетики Венского медицинского университета
Währinger Str.10
AT-1090 Вена (Австрия)
Электронная почта [email protected]
Подробности статьи / публикации
Предварительный просмотр первой страницы
Принята к печати: 27 августа 2018 г.
Опубликована онлайн: 13 ноября 2018 г.
Дата выпуска: январь 2019 г.
Количество страниц для печати: 8
Количество рисунков: 2
Количество столов: 2
ISSN: 1661-8769 (печатный)
eISSN: 1661-8777 (онлайн)
Для дополнительной информации: https: // www.karger.com/MSY
Авторские права / Дозировка препарата / Заявление об ограничении ответственности
Авторские права: Все права защищены. Никакая часть данной публикации не может быть переведена на другие языки, воспроизведена или использована в любой форме или любыми средствами, электронными или механическими, включая фотокопирование, запись, микрокопирование, или с помощью какой-либо системы хранения и поиска информации, без письменного разрешения издателя. .
Дозировка лекарства: авторы и издатель приложили все усилия, чтобы гарантировать, что выбор и дозировка лекарства, указанные в этом тексте, соответствуют текущим рекомендациям и практике на момент публикации.Тем не менее, ввиду продолжающихся исследований, изменений в правительственных постановлениях и постоянного потока информации, касающейся лекарственной терапии и реакций на них, читателю настоятельно рекомендуется проверять листок-вкладыш для каждого препарата на предмет любых изменений показаний и дозировки, а также дополнительных предупреждений. и меры предосторожности. Это особенно важно, когда рекомендованным агентом является новый и / или редко применяемый препарат.
Отказ от ответственности: утверждения, мнения и данные, содержащиеся в этой публикации, принадлежат исключительно отдельным авторам и соавторам, а не издателям и редакторам.Появление в публикации рекламы и / или ссылок на продукты не является гарантией, одобрением или одобрением рекламируемых продуктов или услуг или их эффективности, качества или безопасности. Издатель и редактор (-ы) не несут ответственности за любой ущерб, причиненный людям или имуществу в результате любых идей, методов, инструкций или продуктов, упомянутых в контенте или рекламе.
Артрогрипоз: основы практики, патофизиология, эпидемиология
Taricco LD, Aoki SS.Реабилитация взрослого пациента с врожденным множественным артрогрипозом, получавшего внешний фиксатор. Am J Phys Med Rehabil . 2009 Май. 88 (5): 431-4. [Медлайн].
Бамшад М., Ван Хест А.Е., Удовольствие Д. Артрогрипоз: обзор и обновление. Хирургия костного сустава J Am . 2009 июль 91 Дополнение 4: 40-6. [Медлайн]. [Полный текст].
Холл JG. Генетические аспекты артрогрипоза. Клин Ортоп . 1985. 194: 44-53. [Медлайн].
Дарин Н., Кимбер Э., Кроксмарк А.К., Тулиниус М. Множественные врожденные контрактуры: распространенность при рождении, этиология и исход. Педиатр Дж. . 2002, январь 140 (1): 61-7. [Медлайн].
Лайтинен О., Хирвенсало М. Врожденный множественный артрогрипоз. Энн Педиатр Фенн . 1966. 12: 133-138.
Li Y, Sheng F, Xia C, Xu L, Qiu Y, Zhu Z. Факторы риска нарушения функции легких у пациентов с множественным врожденным артрогрипозом и сопутствующим сколиозом: сравнение с идиопатическим сколиозом у подростков. Позвоночник (Фила Па, 1976) . 2018 15 апреля. 43 (8): E456-60. [Медлайн].
Чирилло А., Коллинз Дж., Савацки Б., Хамди Р., Дахан-Олиэль Н. Боль среди детей и взрослых, живущих с врожденным множественным артрогрипозом: обзорный обзор. Ам Дж Мед Генет С Семин Мед Генет . 2019 Сентябрь 181 (3): 436-53. [Медлайн].
Bevan WP, Hall JG, Bamshad M, Staheli LT, Jaffe KM, Song K. Врожденный множественный артрогрипоз (амиоплазия): ортопедическая перспектива. Дж Педиатр Ортоп . 2007 июль-август. 27 (5): 594-600. [Медлайн].
Alves PV, Zhao L, Patel PK, Bolognese AM. Артрогрипоз: диагностика и планирование лечения пациентов, обращающихся за ортодонтическим лечением или ортогнатической операцией. Дж. Краниофак Сург . 2007 июля 18 (4): 838-43. [Медлайн].
Наркис Г, Ландау Д, Усадьба Е, Офир Р, Бирк О.С. Генетика артрогрипоза: подход к анализу сцепления. Клин Ортоп Релат Рес .2006 28 декабря. [Medline].
Наркис Г., Офир Р., Ландау Д., Манор Е., Волокита М., Гершковиц Р. Летальный контрактный синдром 3 типа (LCCS3) вызывается мутацией в PIP5K1C, который кодирует PIPKI гамма пути фосфатидилинситола. Ам Дж. Хам Генет . 2007 сентябрь 81 (3): 530-9. [Медлайн].
Wallach E, Walther-Louvier U, Espil-Taris C, Rivier F, Baudou E, Cances C. Артрогрипоз у детей: этиологические оценки и подготовка протокола этиологических исследований. Арх. Педиатр . 2018 15 июня. [Medline].
Холл JG. Врожденный множественный артрогрипоз: этиология, генетика, классификация, диагностический подход и общие аспекты. J Педиатр Ортоп B . 1997 июл.6 (3): 159-66. [Медлайн].
Dai S, Dieterich K, Jaeger M, Wuyam B, Jouk PS, Perennou D. Инвалидность у взрослых с артрогрипозом тяжелая, частично незаметная и зависит от генотипа. Неврология . 2018 1 мая.90 (18): e1596-e1604. [Медлайн].
Bamshad M, Jorde LB, Carey JC. Пересмотренная и расширенная классификация дистальных артрогрипозов. Ам Дж. Мед Генет . 1996 11 ноября. 65 (4): 277-81. [Медлайн].
Чен Х. Синдром множественного птеригиума. Атлас генетической диагностики и консультирования . 2-й. Нью-Йорк Дордрехт Гейдельберг Лондон: Спрингер; 2012. 2: 1479-1485.
Чен Х, Чанг СН, Мисра РП.Синдром множественного птеригиума. Ам Дж. Мед Генет . 1980. 7 (2): 91-102. [Медлайн].
Эскобар В., Бикслер Д., Глейзер С., Уивер Д. Д., Гиббс Т. Синдром множественного птеригиума. Ам Дж. Дис Детский . 1978 июн.132 (6): 609-11. [Медлайн].
Холл JG. Летальные множественные синдромы птеригиума. Ам Дж. Мед Генет . 1984, 17 апреля (4): 803-7. [Медлайн].
Холл Дж. Г., Рид С. Д., Грин Г. Дистальные артрогрипозы: определение новых образований — обзор и нозологическое обсуждение. Ам Дж. Мед Генет . 1982, 11 февраля (2): 185-239. [Медлайн].
Entezami M, Runkel S, Kunze J, Weitzel HK, Becker R. Пренатальная диагностика летального синдрома множественного птеригиума типа II. История болезни. Диагностика плода Ther . 1998 Янв-Фев. 13 (1): 35-8. [Медлайн].
Чен Х. Последовательность акинезии плода. Атлас генетической диагностики и консультирования . 2-й. Нью-Йорк Дордрехт Гейдельберг Лондон: Спрингер; 2012. 2: 809-821.
Чен Х, Иммкен Л., Лахман Р. Синдром множественной птеригии, камптодактилии, лицевых аномалий, гипоплазии легких и сердца, кистозной гигромы и аномалий скелета: определение новой сущности и обзор летальных форм синдрома множественного птеригиума. Ам Дж. Мед Генет . 1984 г., 17 (4): 809-26. [Медлайн].
Синдром Чена Х. Фримена-Шелдона. Атлас генетической диагностики и консультирования . 2-й. Нью-Йорк Дордрехт Гейдельберг Лондон: Спрингер; 2012 г.2: 883-889.
Wilnai Y, Seaver LH, Enns GM. Атипичная врожденная амиоплазия у младенца с синдромом Ли: митохондриальная причина тяжелых контрактур ?. Ам Дж. Мед Генет А . 2012 сентябрь 158A (9): 2353-7. [Медлайн].
Чен Х., Блумберг Б., Иммкен Л. Синдром Пена-Шокейра: отчет о пяти случаях и дальнейшее описание синдрома. Ам Дж. Мед Генет . 1983 16 октября (2): 213-24. [Медлайн].
Чен Х, Блэкберн В. Р., Вертелеки В.Акинезия плода и множественные перинатальные переломы. Ам Дж. Мед Генет . 1995 13 февраля. 55 (4): 472-7. [Медлайн].
Холл JG. Анализ фенотипа Pena Shokeir. Ам Дж. Мед Генет . 1986 Сентябрь 25 (1): 99-117. [Медлайн].
Dicke JM, Piper SL, Goldfarb CA. Использование ультразвука для выявления аномалий конечностей плода — 20-летний опыт работы в одном центре. Пренат Диагностика . 2014 4 декабря [Medline].
Каток БД.Артрогрипоз: обзор и подход к пренатальной диагностике. Наблюдение за акушерством и гинекологом . 2011 июн.66 (6): 369-77. [Медлайн].
Dimitraki M, Tsikouras P, Bouchlariotou S, Dafopoulos A, Konstantou E, Liberis V. Пренатальная оценка артрогрипоза. Обзор литературы. J Matern Fetal Neonatal Med . 2011 24 января (1): 32-6. [Медлайн].
Паккасъярви Н., Ритванен А., Херва Р., Пелтонен Л., Кестиля М., Игнатиус Дж. Синдром врожденной смертельной контрактуры (СПКК) и другие летальные артрогрипозы в Финляндии — эпидемиологическое исследование. Ам Дж. Мед Генет А . 2006 г. 1. 140A (17): 1834-9. [Медлайн].
Линь И.В., Чуэ Х.Й., Чанг С.Д., Ченг П.Дж. Применение трехмерной ультрасонографии в пренатальной диагностике артрогрипоза. Тайвань Дж. Обстет Гинеколь . 2008 Март 47 (1): 75-8. [Медлайн].
Ajayi RA, Keen CE, Knott PD. Ультразвуковая диагностика фенотипа Pena Shokeir на 14 неделе беременности. Пренат Диагностика . 1995 15 августа (8): 762-4.[Медлайн].
Скотт Х., Хантер А., Бедар Б. Несмертельный врожденный множественный артрогрипоз с кистозной гигромой на сроке гестации 13 недель. Пренат Диагностика . 1999 октября 19 (10): 966-71. [Медлайн].
Мерфи Дж. К., Нил Д., Бромли Б., Бенасерраф Б. Р., Копель Дж. А. Гипоэхогенность длинных костей плода: новый ультразвуковой маркер артрогрипоза. Пренат Диагностика . 2002 22 декабря (13): 1219-22. [Медлайн].
Hamdy RC, van Bosse H, Altiok H и др.Лечение и исходы артрогрипоза нижней конечности. Ам Дж Мед Генет С Семин Мед Генет . 2019 Сентябрь 181 (3): 372-84. [Медлайн]. [Полный текст].
Gagnon M, Caporuscio K, Veilleux LN, Hamdy R, Dahan-Oliel N. Функции мышц и суставов у детей, живущих с врожденным множественным артрогрипозом: обзорный обзор. Ам Дж Мед Генет С Семин Мед Генет . 2019 Сентябрь 181 (3): 410-26. [Медлайн].
Moessinger AC. Последовательность деформации акинезии плода: модель на животных. Педиатрия . 1983 декабрь 72 (6): 857-63. [Медлайн].
Staheli LT, Hall JG, Jaffe K, et al. Артрогрипоз: Атлас текстов . Нью-Йорк: издательство Кембриджского университета; 1998.
Sawatzky B, Jones T, Miller R, Noureai H. Взаимосвязь между операцией на суставах и качеством жизни у взрослых с артрогрипозом: международное исследование. Ам Дж Мед Генет С Семин Мед Генет . 2019 Сентябрь 181 (3): 469-73. [Медлайн].
Холл JG.Артрогрипоз (множественные врожденные контрактуры): диагностический подход к этиологии, классификации, генетике и общим принципам. евро J Med Genet . 2014 августа 57 (8): 464-72. [Медлайн].
Navti OB, Kinning E, Vasudevan P, Barrow M, Porter H, Howarth E. Обзор перинатального лечения артрогрипоза в большой клинической больнице Великобритании, обслуживающей полиэтническое население. Пренат Диагностика . 2010 30 января (1): 49-56. [Медлайн].
Чен Х.Врожденный множественный артрогрипоз. Атлас генетической диагностики и консультирования . 2-й. Нью-Йорк Дордрехт Гейдельберг Лондон: Спрингер; 2012. 1: 141-156.
Isaacson G, Барабан ET. Трудности в обеспечении проходимости дыхательных путей у детей и молодых людей с артрогрипозом. World J Otorhinolaryngol Head Neck Surg . 2018 июн. 4 (2): 122-5. [Медлайн]. [Полный текст].
[Директива] Michaud LJ. Назначение терапевтических услуг детям с ограниченными двигательными возможностями. Педиатрия . 2004 июн.113 (6): 1836-8. [Медлайн].
фургон Боссе HJP. Ортопедическая помощь ребенку с артрогрипозом: обзор за 2020 год. Curr Opin Педиатр . 2019 16 ноября. [Medline].
Nouraei H, Sawatzky B, MacGillivray M, Hall J. Долгосрочные функциональные и подвижные результаты для людей с врожденным множественным артрогрипозом. Ам Дж. Мед Генет А . 2017 май. 173 (5): 1270-8. [Медлайн].
Синдром дистального артрогрипоза | Энциклопедия.com
Определение
Синдром дистального артрогрипоза — это редкое генетическое заболевание, при котором люди рождаются с характерным изгибом в суставах рук и ног. Контрактура — это слово, используемое для описания того, что происходит в суставах, что вызывает сгибание. Помимо контрактур кисти и стопы, люди с дистальным артрогрипозом рождаются с плотно сжатым кулаком и перекрытием пальцев.
Описание
Слово артрогрипоз означает изогнутый (изогнутый) или искривленный сустав.Дистальный означает наиболее удаленный от какой-либо одной точки отсчета или чего-то удаленного. Таким образом, синдром дистального артрогрипоза вызывает сгибание суставов в самых удаленных частях наших конечностей, кистях и стопах.
Постоянные движения плода во время беременности необходимы для развития суставов. Без регулярных движений суставы становятся тугими, что приводит к контрактурам. Первые случаи артриогрипоза были выявлены в 1923 году. Множественный врожденный артриогрипоз (AMC) также называют последовательностью акинезии / гипокинезии плода, которая не является нарушением, но описывает, что происходит, когда плод не двигается во время развития плода.Причины отсутствия движения плода включают неврологические, мышечные, соединительнотканные или скелетные аномалии или внутриутробную скученность. Существуют различные заболевания, которые включают ту или иную форму артрогрипоза.
Дистальный артрогрипоз был идентифицирован как отдельное генетическое заболевание в 1982 году. Было идентифицировано два типа дистального артрогрипоза. Тип 1 или типичный дистальный артрогрипоз используется для описания людей с дистальными контрактурами кистей и стоп, характерным положением кистей и стоп и нормальным интеллектом.Дистальный артрогрипоз 2 типа известен как атипичная форма. Он характеризуется дополнительными врожденными дефектами и умеренной задержкой умственного развития.
Существуют и другие синдромы, которые включают артрогрипоз, однако дистальный артрогрипоз был охарактеризован как отдельный синдром по его типу наследования . Помимо наследования, есть и другие особенности, которые отличают этот тип артрогрипоза от других форм. Некоторые из этих особенностей включают характерное положение рук при рождении; кулаки сжаты, пальцы согнуты внахлест.Кроме того, у этих людей часто наблюдаются проблемы с положением стопы, называемые косолапостью . Другой отличительной особенностью является чрезвычайно широкая вариабельность тяжести и количества контрактур суставов, которые могут проявляться. Эта вариабельность часто наблюдается между двумя пораженными людьми из одной семьи.
Генетический профиль
Синдром дистального артрогрипоза наследуется по аутосомно-доминантному типу. Паттерны аутосомно-доминантного наследования требуют только одной генетической мутации в одной из пар хромосом для проявления симптомов заболевания. Хромосомы — это структуры, несущие гены. Гены — это образец того, кто мы есть и как мы выглядим. У людей 23 пары или 46 хромосом в каждой клетке своего тела. Первые 22 хромосомы пронумерованы 1-22 и называются аутосомами. Оставшейся паре присваивается буква X или Y, и они являются хромосомами, определяющими пол. Типичный мужчина описывается как 46, XY. Типичной женщине 46 лет ХХ.
Каждый родитель передает своим детям одну из парных хромосом.Перед оплодотворением отцовская сперматозоид делится пополам, и общее количество хромосом сокращается с 46 до 23. Материнская яйцеклетка также подвергается редукции того же типа. На
Во время зачатия каждый родитель передает своим детям 23 хромосомы, по одной из каждой пары. Вся генетическая информация содержится на каждой хромосоме.
Если отец или мать поражены дистальным артрогрипозом, существует 50% -ная вероятность, что они передадут хромосому с геном для этого заболевания каждому из своих детей.Специфический ген дистального артрогрипоза неизвестен, однако мы знаем, что он расположен на хромосоме номер 9.
Симптомы дистального артрогрипоза могут быть разными у двух пораженных родственников. Например, у матери могут быть контрактуры всех суставов, а у ее ребенка контрактуры могут быть только в руках. Из-за такой вариабельности симптомов этого заболевания считается, что существует более одной мутации гена , которая вызывает дистальный артрогрипоз.По состоянию на 2001 год единственный ген, который, как считается, вызывает это заболевание, находится на хромосоме номер 9. Точное местоположение и тип генетической мутации на хромосоме 9 неизвестны, и поэтому единственное генетическое тестирование , доступное по состоянию на 2001 год, основано на исследованиях.
Демография
Дистальный артрогрипоз может поражать людей из всех типов населения и этнических групп. Это заболевание может поражать как мужчин, так и женщин. Было описано лишь несколько человек с этим типом артрогрипоза.Врач, доктор Холл, назвавший это заболевание в 1982 году, первоначально идентифицировал 37 пациентов с синдромом дистального артрогрипоза 1 и 2 типа. Она выявила 14 человек с типом 1 и 23 человека с типом 2. С тех пор у многих других людей был диагностирован дистальный артрогрипоз. Точная заболеваемость в литературе не сообщается.
Признаки и симптомы
При рождении многим людям ставят диагноз на основании их характерного положения рук.Практически все люди с дистальным артрогрипозом рождаются со сжатыми руками в кулак. Большой палец повернут внутрь, лежа на ладони, это называется отведением. Пальцы также накладываются друг на друга. Такое положение руки также характерно для более серьезного состояния, называемого трисомией 18 . У большинства пациентов с дистальным артрогрипозом также возникают проблемы с положением стопы. Многие пациенты страдают косолапостью в той или иной форме, когда ступня искажается по форме или положению.Еще одно слово для обозначения косолапости — косолапость.
Помимо поражения кисти и стопы, у небольшого процента пациентов будет вывих или разделение тазобедренного сустава, а также трудности с сгибанием бедра и тенденция к небольшому неестественному сгибанию в тазобедренных суставах. . Колени также могут иметь аналогичные проблемы из-за того, что они слегка согнуты и зафиксированы в этой точке. Немногие люди рождаются с жесткими плечами.
Синдром дистального артрогрипоза 2-го типа включает другие врожденные дефекты, не наблюдаемые у лиц 1-го типа.Например, дистальный артрогрипоз 2 типа включает проблемы с закрытием губы, называемые заячьей губой, или отверстие в небе, называемое расщелиной неба.
Другие аномалии, наблюдаемые при дистальном артрогрипозе 2 типа, включают маленький язык, низкий рост, искривление позвоночника, более серьезные контрактуры суставов и задержку умственного развития.
Диагноз
Диагноз дистального артрогрипоза иногда может быть поставлен во время беременности на основании ультразвукового исследования. Ультразвук может выявить характерные признаки кисти, а также деформации сгибания рук и ног.Пораженный плод может испытывать затруднения при глотании, и это проявляется в ультразвуковой оценке в виде дополнительных околоплодных вод, окружающих ребенка, которые называются многоводием. Еще один очень важный и специфический диагностический признак дистального артрогрипоза во время беременности — отсутствие движений плода. На УЗИ уже на 17 неделе беременности.
После рождения диагноз ставит врач, проводящий физический осмотр ребенка с подозрением на это заболевание.Если у ребенка дистальный артрогрипоз 2 типа, ему может быть трудно правильно есть. С 2001 года единственный доступный тип генетического тестирования основан на исследованиях. Поскольку, вероятно, существует более одного гена, вызывающего заболевание, генетическое тестирование, проводимое в настоящее время, еще не предлагается больным людям для подтверждения диагноза.
Лечение и ведение
Лечение пациентов с дистальным артрогрипозом адаптировано к потребностям пострадавшего ребенка.С терапией после родов, которая помогает расслабить суставы и повторно тренировать мышцы, большинство людей чувствуют себя хорошо. Руки не остаются сжатыми всю жизнь, но в конечном итоге разжимаются. Иногда пальцы остаются в некоторой степени согнутыми. Косолапость обычно можно исправить, сделав стопы прямыми.
Прогноз
Прогноз зависит от того, насколько серьезно поражен человек и сколько суставов поражено. Некоторые из более тяжелых случаев могут быть связаны с ранней смертью из-за внезапной дыхательной недостаточности и затрудненного дыхания.Большинство людей с дистальным артрогрипозом очень хорошо себя чувствуют после получения необходимого лечения, а иногда и хирургического вмешательства по исправлению тяжелых сокращений суставов.
Ресурсы
КНИГИ
Fleischer, A., et al. Сонография в акушерстве и гинекологии, принципы и практика. Стэмфорд, Коннектикут: 1996.
Джонс, Кеннет. Распознаваемые модели пороков развития человека Смита. 5-е изд. Филадельфия: W.B. Saunders Company, 1997.
PERIODICALS
Sonoda, T.«Два брата с дистальным артрогрипозом, своеобразной внешностью лица, волчьей пастью, низким ростом, гидронефрозом, ретенцией яичек и нормальным интеллектом: новый тип дистального артрогрипоза?» Американский журнал медицинской генетики.