Врожденная косолапость мкб 10: Ошибка 404. Файл не найден

Q66 Врожденные деформации стопы…
- Q66.0 Конско-варусная косолапость
- Q66.1 Пяточно-варусная косолапость
- Q66.2 Варусная стопа
- Q66.
 3 Другие врожденные варусные деформации стопы
3 Другие врожденные варусные деформации стопы - Q66.4 Пяточно-вальгусная косолапость
- Q66.5 Врожденная плоская стопа [pes planus]
- Q66.
 6 Другие врожденные вальгусные деформации стопы
6 Другие врожденные вальгусные деформации стопы - Q66.7 Полая стопа [pes cavus]
- Q66.8 Другие врожденные деформации стопы
- Q66.
 9 Врожденная деформация стопы неуточненная
9 Врожденная деформация стопы неуточненная
 3 Другие врожденные варусные деформации стопы
3 Другие врожденные варусные деформации стопы- Выбор препаратов
Подобрать препарат можно с помощью фильтров. Чтобы увидеть в перечне лекарства, входящие в подгруппы, отметьте галочкой «включить препараты подгрупп». Нажав на иконку , можно добавить препарат в избранное и проверить на дубли и межлекарственные взаимодействия.
Полужирным начертанием выделены лекарства, входящие в справочники текущего года. Рядом с названием препарата может быть указан еженедельный уровень индекса информационного спроса (показатель, который отражает степень интереса потребителей к информации о лекарстве).
Сбросить фильтры
включить препараты подгрупп
Войти через:
МКБ-10 Класс XVII. Врожденные аномалии (пороки развития), деформации и хромосомные нарушения • Медицинский Клуб
- Q00-Q07 — Врожденные аномалии (пороки развития) нервной системы
- Q10-Q18 — Врожденные аномалии (пороки развития) глаза, уха, лица и шеи
- Q20-Q28 — Врожденные аномалии (пороки развития) системы кровообращения
- Q30-Q34 — Врожденные аномалии (пороки развития) органов дыхания
- Q35-Q37 — Расщелина губы и неба (заячья губа и волчья пасть)
- Q38-Q45 — Другие врожденные аномалии (пороки развития) органов пищеварения
- Q50-Q56 — Врожденные аномалии (пороки) половых органов
- Q60-Q64 — Врожденные аномалии (пороки развития) мочевой системы
- Q65-Q79 — Врожденные аномалии (пороки развития) и деформации костно-мышечной системы
- Q80-Q89 — Другие врожденные аномалии (пороки развития)
- Q90-Q99 — Хромосомные аномалии, не классифицированные в других рубриках
Q00-Q07.
 Врожденные аномалии (пороки развития) нервной системы
Врожденные аномалии (пороки развития) нервной системы
Q00. Анэнцефалия и подобные пороки развития
- Q00.0. Анэнцефалия
- Q00.1. Краниорахишизис
- Q00.2. Инэнцефалия
Q01. Энцефалоцеле
- Q01.0. Лобное энцефалоцеле
- Q01.1. Носолобное энцефалоцеле
- Q01.2. Затылочное энцефалоцеле
- Q01.8. Энцефалоцеле других областей
- Q01.9. Энцефалоцеле неуточненное
Q02. Микроцефалия
Q03. Врожденная гидроцефалия
- Q03.0. Врожденный порок сильвиева водопровода
- Q03.1. Атрезия отверстий Мажанди и Лушки
- Q03.8. Другая врожденная гидроцефалия
- Q03.9. Врожденная гидроцефалия неуточненная
Q04. Другие врожденные аномалии (пороки развития) мозга
- Q04.0. Врожденная аномалия мозолистого тела
- Q04.1. Аринэнцефалия
- Q04.2. Голопрозэнцефалия
- Q04.3. Другие редукционные деформации мозга
- Q04.4. Сентооптическая дисплазия
- Q04.
 5. Мегалэнцефалия
5. Мегалэнцефалия - Q04.6. Врожденные церебральные кисты
- Q04.8. Другие уточненные врожденные аномалии мозга
- Q04.9. Врожденная аномалия мозга неуточненная
Q05. Spina bifida (неполное закрытие позвоночного канала)
- Q05.0. Spina bifida в шейном отделе с гидроцефалией
- Q05.1. Spina bifida в грудном отделе с гидроцефалией
- Q05.2. Spina bifida в поясничном отделе с гидроцефалией
- Q05.3. Spina bifida в сакральном отделе с гидроцефалией
- Q05.4. Spina bifida с гидроцефалией неуточненная
- Q05.5. Spina bifida в шейном отделе без гидроцефалии
- Q05.6. Spina bifida в грудном отделе без гидроцефалии
- Q05.7. Spina bifida в поясничном отделе без гидроцефалии
- Q05.8. Spina bifida в крестцовом отделе БДУ
- Q05.9. Spina bifida неуточненная
Q06. Другие врожденные аномалии (пороки развития) спинного мозга
- Q06.0. Амиелия
- Q06.1. Гипоплазия и дисплазия спинного мозга
- Q06.
 2. Диастематомиелия
2. Диастематомиелия - Q06.3. Другие пороки развития конского хвоста
- Q06.4. Гидромиелия
- Q06.8. Другие уточненные пороки развития спинного мозга
- Q06.9. Врожденный порок развития спинного мозга неуточненный
Q07. Другие врожденные аномалии (пороки развития) нервной системы
- Q07.0. Синдром Арнольда-Киари
- Q07.8. Другие уточненные пороки развития нервной системы
- Q07.9. Порок развития нервной системы неуточненный
Q10-Q18. Врожденные аномалии (пороки развития) глаза, уха, лица и шеи
Q10. Врожденные аномалии (пороки развития) века, слезного аппарата и глазницы
- Q10.0. Врожденный птоз
- Q10.1. Врожденный эктропион
- Q10.2. Врожденный энтропион
- Q10.3. Другие пороки развития века
- Q10.4. Отсутствие или агенезия слезного аппарата
- Q10.5. Врожденные стеноз и стриктура слезного протока
- Q10.6. Другие пороки развития слезного аппарата
- Q10.
 7. Порок развития глазницы
7. Порок развития глазницы
Q11. Анофтальм, микрофтальм и макрофтальм
- Q11.0. Киста глазного яблока
- Q11.1. Другой вид анофтальма
- Q11.2. Микрофтальм
- Q11.3. Макрофтальм
Q12. Врожденные аномалии (пороки развития) хрусталика
- Q12.0. Врожденная катаракта
- Q12.1. Врожденное смещение хрусталика
- Q12.2. Колобома хрусталика
- Q12.3. Врожденная афакия
- Q12.4. Сферофакия
- Q12.8. Другие врожденные пороки хрусталика
- Q12.9. Врожденный порок хрусталика неуточненный
Q13. Врожденные аномалии (пороки развития) переднего сегмента глаза
- Q13.0. Колобома радужки
- Q13.1. Отсутствие ражужки
- Q13.2. Другие пороки развития радужки
- Q13.3. Врожденное помутнение роговицы
- Q13.4. Другие пороки развития роговицы
- Q13.5. Голубая склера
- Q13.8. Другие врожденные аномалии переднего сегмента глаза
- Q13.9. Врожденная аномалия переднего сегмента глаза неуточненная
Q14.
 Врожденные аномалии (пороки развития) заднего сегмента глаза
Врожденные аномалии (пороки развития) заднего сегмента глаза
- Q14.0. Врожденная аномалия стекловидного тела
- Q14.1. Врожденная аномалия сетчатки
- Q14.2. Врожденная аномалия диска зрительного нерва
- Q14.3. Врожденная аномалия сосудистой оболочки глаза
- Q14.8. Другие врожденные аномалии заднего сегмента глаза
- Q14.9. Врожденная аномалия заднего сегмента глаза неуточненная
Q15. Другие врожденные аномалии (пороки развития) глаза
- Q15.0. Врожденная глаукома
- Q15.8. Другие уточненные пороки развития глаза
- Q15.9. Врожденный порок глаза неуточненный
Q16. Врожденные аномалии (пороки развития) уха, вызывающие нарушение слуха
- Q16.0. Врожденное отсутствие ушной раковины
- Q16.1. Врожденное отсутствие, атрезия и стриктура слухового прохода (наружного)
- Q16.2. Отсутствие евстахиевой трубы
- Q16.3. Врожденная аномалия слуховых косточек
- Q16.4. Другие врожденные аномалии среднего уха
- Q16.
 5. Врожденная аномалия внутреннего уха
5. Врожденная аномалия внутреннего уха - Q16.9. Врожденная аномалия уха, вызывающая нарушение слуха, неуточненная
Q17. Другие врожденные аномалии (пороки развития) уха
- Q17.0. Добавочная ушная раковина
- Q17.1. Макротия
- Q17.2. Микротия
- Q17.3. Другая аномалия уха
- Q17.4. Аномально расположенное ухо
- Q17.5. Выступающее ухо
- Q17.8. Другие уточненные пороки развития уха
- Q17.9. Порок развития уха неуточненный
Q18. Другие врожденные аномалии (пороки развития) лица и шеи
- Q18.0. Пазуха, фистула и киста жаберной щели
- Q18.1. Преаурикулярная пазуха и киста
- Q18.2. Другие пороки развития жаберной щели
- Q18.3. Крыловидная шея
- Q18.4. Макростомия
- Q18.5. Микростомия
- Q18.6. Макрохейлия
- Q18.7. Микрохейлия
- Q18.8. Другие уточненные пороки развития лица и шеи
- Q18.9. Порок развития лица и шеи неуточненный
Q20-Q28.
 Врожденные аномалии (пороки развития) системы кровообращения
Врожденные аномалии (пороки развития) системы кровообращения
Q20. Врожденные аномалии (пороки развития) сердечных камер и соединений
- Q20.0. Общий артериальный ствол
- Q20.1. Удвоение выходного отверстия правого желудочка
- Q20.2. Удвоение выходного отверстия левого желудочка
- Q20.3. Дискордантное желудочково-артериальное соединение
- Q20.4. Удвоение входного отверстия желудочка
- Q20.5. Дискордантное предсердно-желудочковое соединение
- Q20.6. Изомерия ушка предсердия
- Q20.8. Другие врожденные аномалии сердечных камер и соединений
- Q20.9. Врожденная аномалия сердечных камер и соединений неуточненная
Q21. Врожденные аномалии (пороки развития) сердечной перегородки
- Q21.0. Дефект межжелудочковой перегородки
- Q21.1. Дефект предсердной перегородки
- Q21.2. Дефект предсердно-желудочковой перегородки
- Q21.3. Тетрада Фалло
- Q21.4. Дефектперегородки между аортой и легочной артерией
- Q21.
 8. Другие врожденные аномалии сердечной перегородки
8. Другие врожденные аномалии сердечной перегородки - Q21.9. Врожденная аномалия сердечной перегородки неуточненная
Q22. Врожденные аномалии (пороки развития) легочного и трехстворчатого клапанов
- Q22.0. Атрезия клапана легочной артерии
- Q22.1. Врожденный стеноз клапана легочной артерии
- Q22.2. Врожденная недостаточность клапана легочной артерии
- Q22.3. Другие врожденные пороки клапана легочной артерии
- Q22.4. Врожденный стеноз трехстворчатого клапана
- Q22.5. Аномалия Эбштейна
- Q22.6. Синдром правосторонней гипоплазии сердца
- Q22.8. Другие врожденные аномалии трехстворчатого клапана
- Q22.9. Врожденная аномалия трехстворчатого клапана неуточненная
Q23. Врожденные аномалии (пороки развития) аортального и митрального клапанов
- Q23.0. Врожденный стеноз аортального клапана
- Q23.1. Врожденная недостаточность аортального клапана
- Q23.2. Врожденный митральный стеноз
- Q23.
 3. Врожденная митральная недостаточность
3. Врожденная митральная недостаточность - Q23.4. Синдром левосторонней гипоплазии сердца
- Q23.8. Другие врожденные аномалии аортального и митрального клапанов
- Q23.9. Врожденная аномалия аортального и митрального клапанов неуточненная
Q24. Другие врожденные аномалии (пороки развития) сердца
- Q24.0. Декстрокардия
- Q24.1. Левокардия
- Q24.2. Трехпредсердное сердце
- Q24.3. Воронкообразный стеноз клапана легочной артерии
- Q24.4. Врожденный субаортальный стеноз
- Q24.5. Аномалия развития коронарных сосудов
- Q24.6. Врожденная сердечная блокада
- Q24.8. Другие уточненные врожденные аномалии сердца
- Q24.9. Врожденный порок сердца неуточненный
Q25. Врожденные аномалии (пороки развития) крупных артерий
- Q25.0. Открытый артериальный проток
- Q25.1. Коарктация аорты
- Q25.2. Атрезия аорты
- Q25.3. Стеноз аорты
- Q25.4. Другие врожденные аномалии аорты
- Q25.
 5. Атрезия легочной артерии
5. Атрезия легочной артерии - Q25.6. Стеноз легочной артерии
- Q25.7. Другие врожденные аномалии легочной артерии
- Q25.8. Другие врожденные аномалии крупных артерий
- Q25.9. Врожденная аномалия крупных артерий неуточненная
Q26. Врожденные аномалии (пороки развития) крупных вен
- Q26.0. Врожденный стеноз полой вены
- Q26.1. Сохранение левой верхней полой вены
- Q26.2. Тотальная аномалия соединения легочных вен
- Q26.3. Частичная аномалия соединения легочных вен
- Q26.4. Аномалия соединения легочных вен неуточненная
- Q26.5. Аномалия соединения портальной вены
- Q26.6. Портальная венозно-печеночно-артериальная фистула
- Q26.8. Другие врожденные аномалии крупных вен
- Q26.9. Порок развития крупной вены неуточненный
Q27. Другие врожденные аномалии (пороки развития) системы периферических сосудов
- Q27.0. Врожденное отсутствие и гипоплазия пупочной артерии
- Q27.
 1. Врожденный стеноз почечной артерии
1. Врожденный стеноз почечной артерии - Q27.2. Другие пороки развития почечной артерии
- Q27.3. Периферический артериовенозный порок развития
- Q27.4. Врожденная флебэктазия
- Q27.8. Другие уточненные врожденные аномалии системы периферических сосудов
- Q27.9. Врожденная аномалия системы периферических сосудов неуточненная
Q28. Другие врожденные аномалии (пороки развития) системы кровообращения
- Q28.0. Артериовенозная аномалия развития прецеребральных сосудов
- Q28.1. Другие пороки развития прецеребральных сосудов
- Q28.2. Артериовенозный порок развития церебральных сосудов
- Q28.3. Другие пороки развития церебральных сосудов
- Q28.8. Другие уточненные врожденные аномалии системы кровообращения
- Q28.9. Врожденная аномалия системы кровообращения неуточненная
Q30-Q34. Врожденные аномалии (пороки развития) органов дыхания
Q30. Врожденные аномалии (пороки развития) носа
- Q30.
 0. Атрезия хоан
0. Атрезия хоан - Q30.1. Агенезия и недоразвитие носа
- Q30.2. Треснутый, вдавленный, расщепленный нос
- Q30.3. Врожденная перфорация носовой перегородки
- Q30.8. Другие врожденные аномалии носа
- Q30.9. Врожденная аномалия носа неуточненная
Q31. Врожденные аномалии (пороки развития) гортани
- Q31.0. Перепонка гортани
- Q31.1. Врожденный стеноз гортани под собственно голосовым аппаратом
- Q31.2. Гипоплазия гортани
- Q31.3. Ларингоцеле
- Q31.4. Врожденный стридор гортани
- Q31.8. Другие врожденные пороки развития гортани
- Q31.9. Врожденная аномалия гортани неуточненная
Q32. Врожденные аномалии (пороки развития) трахеи и бронхов
- Q32.0. Врожденная трахеомаляция
- Q32.1. Другие пороки развития трахеи
- Q32.2. Врожденная бронхомаляция
- Q32.3. Врожденный стеноз бронхов
- Q32.4. Другие врожденные аномалии бронхов
Q33. Врожденные аномалии (пороки развития) легкого
- Q33.
 0. Врожденная киста легкого
0. Врожденная киста легкого - Q33.1. Добавочная доля легкого
- Q33.2. Секвестрация легкого
- Q33.3. Агенезия легкого
- Q33.4. Врожденная бронхоэктазия
- Q33.5. Эктопия ткани в легком
- Q33.6. Гипоплазия и дисплазия легкого
- Q33.8. Другие врожденные аномалии легкого
- Q33.9. Врожденная аномалия легкого неуточненная
Q34. Другие врожденные аномалии (пороки развития) органов дыхания
- Q34.0. Аномалия плевры
- Q34.1. Врожденная киста средостения
- Q34.8. Другие уточненные врожденные аномалии органов дыхания
- Q34.9. Врожденная аномалия органов дыхания неуточненная
Q35-Q37. Расщелина губы и неба (заячья губа и волчья пасть)
Q35. Расщелина неба (волчья пасть)
- Q35.0. Расщелина твердого неба двусторонняя
- Q35.1. Расщелина твердого неба односторонняя
- Q35.2. Расщелина мягкого неба двусторонняя
- Q35.3. Расщелина мягкого неба односторонняя
- Q35.
 4. Расщелина твердого и мягкого неба двусторонняя
4. Расщелина твердого и мягкого неба двусторонняя - Q35.5. Расщелина твердого и мягкого неба односторонняя
- Q35.6. Срединная расщелина неба
- Q35.7. Расщелина языка
- Q35.8. Расщелина неба (волчья пасть) неуточненная двусторонняя
- Q35.9. Расщелина неба (волчья пасть) неуточненная односторонняя
Q36. Расщелина губы (заячья губа)
- Q36.0. Расщелина губы двусторонняя
- Q36.1. Расщелина губы срединная
- Q36.9. Расщелина губы односторонняя
Q37. Расщелины неба и губы (волчья пасть с заячьей губой)
- Q37.0. Расщелина твердого неба и губы двусторонняя
- Q37.1. Расщелина твердого неба и губы односторонняя
- Q37.2. Расщелина мягкого неба и губы двусторонняя
- Q37.3. Расщелина мягкого неба и губы односторонняя
- Q37.4. Расщелина твердого и мягкого неба и губы двусторонняя
- Q37.5. Расщелина твердого и мягкого неба и губы односторонняя
- Q37.8. Двусторонняя расщелина неба и губы неуточненная
- Q37.
 9. Односторонняя расщелина неба и губы неуточненная
9. Односторонняя расщелина неба и губы неуточненная
Q38-Q45. Другие врожденные аномалии (пороки развития) органов пищеварения
Q38. Другие врожденные аномалии (пороки развития) языка, рта и глотки
- Q38.0. Врожденные аномалии губ, не классифицированные в других рубриках
- Q38.1. Анкилоглоссия
- Q38.2. Макроглоссия
- Q38.3. Другие врожденные аномалии языка
- Q38.4. Врожденные аномалии слюнных желез
- Q38.5. Врожденные аномалии неба, не классифицированные в других рубриках
- Q38.6. Другие пороки развития рта
- Q38.7. Глоточный карман
- Q38.8. Другие пороки развития глотки
Q39. Врожденные аномалии (пороки развития) пищевода
- Q39.0. Атрезия пищевода без свища
- Q39.1. Атрезия пищевода с трахеально-пищеводным свищом
- Q39.2. Врожденный трахеально-пищеводный свищ без атрезии
- Q39.3. Врожденные стеноз и стриктура пищевода
- Q39.4. Пищеводная перепонка
- Q39.
 5. Врожденное расширение пищевода
5. Врожденное расширение пищевода - Q39.6. Дивертикул пищевода
- Q39.8. Другие врожденные аномалии пищевода
- Q39.9. Врожденная аномалия пищевода неуточненная
Q40. Другие врожденные аномалии (пороки развития) верхней части пищеварительного тракта
- Q40.0. Врожденный гипертрофический пилоростеноз
- Q40.1. Врожденная грыжа пишеводного отверстия диафрагмы
- Q40.2. Другие уточненные пороки развития желудка
- Q40.3. Порок развития желудка неуточненный
- Q40.8. Другие уточненные пороки развития верхней части пищеварительного тракта
- Q40.9. Пороки развития верхней части пищеварительного тракта неуточненные
Q41. Врожденные отсутствие, атрезия и стеноз тонкого кишечника
- Q41.0. Врожденные отсутствие, атрезия и стеноз двенадцатиперстной кишки
- Q41.1. Врожденные отсутствие, атрезия и стеноз тощей кишки
- Q41.2. Врожденные отсутствие, атрезия и стеноз других подвздошной кишки
- Q41.
 8. Врожденные отсутствие атрезия и стеноз других уточненных частей тонкого кишечника
8. Врожденные отсутствие атрезия и стеноз других уточненных частей тонкого кишечника - Q41.9. Врожденные отсутствие, атрезия и стеноз кишечника неуточненной части
Q42. Врожденные отсутствие, атрезия и стеноз толстого кишечника
- Q42.0. Врожденные отсутствие, атрезия и стеноз прямой кишки со свищом
- Q42.1. Врожденные отсутствие, атрезия и стеноз прямой кишки без свища
- Q42.2. Врожденные отсутствие, атрезия и стеноз заднего прохода без свища
- Q42.3. Врожденные отсутствие, атрезия и стеноз заднего прохода без свища
- Q42.8. Врожденные отсутствие, атрезия и стеноз других частей толстого кишечника
- Q42.9. Врожденные отсутствие, атрезия и стеноз толстого кишечника неуточненной части
Q43. Другие врожденные аномалии (пороки развития) кишечника
- Q43.0. Дивертикул Меккеля
- Q43.1. Болезнь Гиршпрунга
- Q43.2. Другие врожденные функциональные аномалии ободочной кишки
- Q43.3. Врожденные аномалии фиксации кишечника
- Q43.
 4. Удвоение кишечника
4. Удвоение кишечника - Q43.5. Эктопический задний проход
- Q43.6. Врожденный свищ прямой кишки и ануса
- Q43.7. Сохранившаяся клоака
- Q43.8. Другие уточненные врожденные аномалии кишечника
- Q43.9. Врожденная аномалия кишечника неуточненная
Q44. Врожденные аномалии (пороки развития) желчного пузыря, желчных протоков и печени
- Q44.0. Агенезия, аплазия и гипоплазия желчного пузыря
- Q44.1. Другие врожденные аномалии желчного пузыря
- Q44.2. Атрезия желчных протоков
- Q44.3. Врожденный стеноз и стриктура желчных протоков
- Q44.4. Киста желчного протока
- Q44.5. Другие врожденные аномалии желчных протоков
- Q44.6. Кистозная болезнь печени
- Q44.7. Другие врожденные аномалии печени
Q45. Другие врожденные аномалии (пороки развития) органов пищеварения
- Q45.0. Агенезия, аплазия и гипоплазия поджелудочной железы
- Q45.1. Кольцевидная поджелудочная железа
- Q45.
 2. Врожденная киста поджелудочной железы
2. Врожденная киста поджелудочной железы - Q45.3. Другие врожденные аномалии поджелудочной железы и протока поджелудочной железы
- Q45.8. Другие уточненные врожденные аномалии органов пищеварения
- Q45.9. Порок развития органов пищеварения неуточненный
Q50-Q56. Врожденные аномалии (пороки) половых органов
Q50. Врожденные аномалии (пороки развития) яичников, фаллопиевых труб и широких связок
- Q50.0. Врожденное отсутствие яичника
- Q50.1. Кистозная аномалия развития яичника
- Q50.2. Врожденный перекрут яичника
- Q50.3. Другие врожденные аномалии яичника
- Q50.4. Эмбриональная киста фаллопиевой трубы
- Q50.5. Эмбриональная киста широкой связки
- Q50.6. Другие врожденные аномалии фаллопиевой трубы и широкой связки
Q51. Врожденные аномалии (пороки развития) тела и шейки матки
- Q51.0. Агенезия и аплазия матки
- Q51.1. Удвоение тела матки с удвоением шейки матки и влагалища
- Q51.
 2. Другие удвоения матки
2. Другие удвоения матки - Q51.3. Двурогая матка
- Q51.4. Однорогая матка
- Q51.5. Агенезия и аплазия шейки матки
- Q51.6. Эмбриональная киста шейки матки
- Q51.7. Врожденный свищ между маткой и пищеварительным и мочевым трактами
- Q51.8. Другие врожденные аномалии тела и шейки матки
- Q51.9. Врожденная аномалия тела и шейки матки неуточненная
Q52. Другие врожденные аномалии (пороки развития) женских половых органов
- Q52.0. Врожденное отсутствие влагалища
- Q52.1. Удвоение влагалища
- Q52.2. Врожденный ректовагинальный свищ
- Q52.3. Девственная плева, полностью закрывающая вход во влагалище
- Q52.4. Другие врожденные аномалии влагалища
- Q52.5. Сращение губ
- Q52.6. Врожденная аномалия клитора
- Q52.7. Другие врожденные аномалии вульвы
- Q52.8. Другие уточненные врожденные аномалии женских половых органов
- Q52.9. Врожденная аномалия женских половых органов неуточненная
Q53.
 Неопущение яичка
Неопущение яичка
- Q53.0. Эктопическое яичко
- Q53.1. Неопущение яичка одностороннее
- Q53.2. Неопущение яичка двустороннее
- Q53.9. Неопущение яичка неуточненное
Q54. Гипоспадия
- Q54.0. Гипоспадия головки полового члена
- Q54.1. Гипоспадия полового члена
- Q54.2. Гипоспадия члено-мошоночная
- Q54.3. Гипоспадия промежностная
- Q54.4. Врожденное искривление полового члена
- Q54.8. Другая гипоспадия
- Q54.9. Гипоспадия неуточненная
Q55. Другие врожденные аномалии (пороки развития) мужских половых органов
- Q55.0. Отсутствие и аплазия яичка
- Q55.1. Гипоплазия яичка и мошонки
- Q55.2. Другие врожденные аномалии яичка и мошонки
- Q55.3. Атрезия семявыносящего протока
- Q55.4. Другие врожденные аномалии семявыносящего протока, придатка яичка, семенного канатика и предстательной железы
- Q55.5. Врожденные отсутствие и аплазия полового члена
- Q55.
 6. Другие врожденные аномалии полового члена
6. Другие врожденные аномалии полового члена - Q55.8. Другие уточненные врожденные аномалии мужских половых органов
- Q55.9. Врожденная аномалия мужских половых органов неуточненная
Q56. Неопределенность пола и псевдогермафродитизм
- Q56.0. Гермафродитизм, не классифицированный в других рубриках
- Q56.1. Мужской псевдогермафродитизм, не классифицированный в других рубриках
- Q56.2. Женский псевдогермафродитизм, не классифицированный в других рубриках
- Q56.3. Псевдогермафродитизм неуточненный
- Q56.4. Неопределенность пола неуточненная
Q60-Q64. Врожденные аномалии (пороки развития) мочевой системы
Q60. Агенезия и другие редукционные дефекты почки
- Q60.0. Агенезия почки односторонняя
- Q60.1. Агенезия почки двусторонняя
- Q60.2. Агенезия почки неуточненная
- Q60.3. Гипоплазия почки односторонняя
- Q60.4. Гипоплазия почки двустронняя
- Q60.5. Гипоплазия почки неуточненная
- Q60.
 6. Синдром Поттера
6. Синдром Поттера
Q61. Кистозная болезнь почек
- Q61.0. Врожденная одиночная киста почки
- Q61.1. Поликистоз почки, детский тип
- Q61.2. Поликистоз почки, взрослый тип
- Q61.3. Поликистоз почки неуточненный
- Q61.4. Дисплазия почки
- Q61.5. Медуллярный кистоз почки
- Q61.8. Другие кистозные болезни почки
- Q61.9. Кистозная болезнь почек неуточненная
Q62. Врожденные нарушения проходимости почечной лоханки и врожденные аномалии мочеточника
- Q62.0. Врожденный гидронефроз
- Q62.1. Атрезия и стеноз мочеточника
- Q62.2. Врожденное расширение мочеточника (врожденный мегалоуретер)
- Q62.3. Другие врожденные нарушения проходимости почечной лоханки и мочеточника
- Q62.4. Агенезия мочеточника
- Q62.5. Удвоение мочеточника
- Q62.6. Неправильное расположение мочеточника
- Q62.7. Врожденный пузырно-мочеточниково-почечный рефлюкс
- Q62.8. Другие врожденные аномалии мочеточника
Q63.
 Другие врожденные аномалии (пороки развития) почки
Другие врожденные аномалии (пороки развития) почки
- Q63.0. Добавочная почка
- Q63.1. Слившаяся, дольчатая и подковообразная почка
- Q63.2. Эктопическая почка
- Q63.3. Гиперпластическая и гигантская почка
- Q63.8. Другие уточненные врожденные аномалии почки
- Q63.9. Врожденная аномалия почки неуточненная
Q64. Другие врожденные аномалии (пороки развития) мочевой системы
- Q64.0. Эписпадия
- Q64.1. Экстрофия мочевого пузыря
- Q64.2. Врожденные задние уретральные клапаны
- Q64.3. Другие виды атрезии и стеноза уретры и шейки мочевого пузыря
- Q64.4. Аномалия мочевого протока (урахуса)
- Q64.5. Врожденное отсутствие мочевого пузыря и мочеиспускательного канала
- Q64.6. Врожденный дивертикул мочевого пузыря
- Q64.7. Другие врожденные аномалии мочевого пузыря и мочеиспускательного канала
- Q64.8. Другие уточненные врожденные аномалии мочевыделительной системы
- Q64.
 9. Врожденная аномалия мочевыделительной системы неуточненная
9. Врожденная аномалия мочевыделительной системы неуточненная
Q65-Q79. Врожденные аномалии (пороки развития) и деформации костно-мышечной системы
Q65. Врожденные деформации бедра
- Q65.0. Врожденный вывих бедра односторонний
- Q65.1. Врожденный вывих бедра двусторонний
- Q65.2. Врожденный вывих бедра неуточненный
- Q65.3. Врожденный подвывих бедра односторонний
- Q65.4. Врожденный подвывих бедра двусторонний
- Q65.5. Врожденный подвывих бедра неуточненный
- Q65.6. Неустойчивое бедро
- Q65.8. Другие врожденные деформации бедра
- Q65.9. Врожденная деформация бедра неуточненная
Q66. Врожденные деформации стопы
- Q66.0. Конско-варусная косолапость
- Q66.1. Пяточно-варусная косолапость
- Q66.2. Варусная стопа
- Q66.3. Другие врожденные варусные деформации
- Q66.4. Пяточно-вальгусная косолапость
- Q66.5. Врожденная плоская стопа (pes planus)
- Q66.
 6. Другие врожденные вальгусные деформации стопы
6. Другие врожденные вальгусные деформации стопы - Q66.7. Полая стопа (pes cavus)
- Q66.8. Другие врожденные деформации стопы
- Q66.9. Врожденная деформация стопы неуточненная
Q67. Врожденные костно-мышечные деформации головы, лица, позвоночника и грудной клетки
- Q67.0. Асимметрия лица
- Q67.1. Сдавленное лицо
- Q67.2. Долихоцефалия
- Q67.3. Плагиоцефалия
- Q67.4. Другие врожденные деформации черепа, лица и челюсти
- Q67.5. Врожденная деформация позвоночника
- Q67.6. Впалая грудь
- Q67.7. Килевидная грудь
- Q67.8. Другие врожденные деформации грудной клетки
Q68. Другие врожденные костно-мышечные деформации
- Q68.0. Врожденная деформация грудиноключично-сосцевидной мышцы
- Q68.1. Врожденная деформация кисти
- Q68.2. Врожденная деформация колена
- Q68.3. Врожденное искривление бедра
- Q68.4. Врожденное искривление большеберцовой и малоберцовой костей
- Q68.
 5. Врожденное искривление длинных костей голени неуточненное
5. Врожденное искривление длинных костей голени неуточненное - Q68.8. Другие уточненные врожденные костно-мышечные деформации
Q69. Полидактилия
- Q69.0. Добавочный палец (пальцы)
- Q69.1. Добавочный большой палец (пальцы) кисти
- Q69.2. Добавочный палец (пальцы) стопы
- Q69.9. Полидактилия неуточненная
Q70. Синдактилия
- Q70.0. Сращение пальцев кисти
- Q70.1. Перепончатость пальцев кисти
- Q70.2. Сращение пальцев стопы
- Q70.3. Перепончатость пальцев стопы
- Q70.4. Полисиндактилия
- Q70.9. Синдактилия неуточненная
Q71. Дефекты, укорачивающие верхнюю конечность
- Q71.0. Врожденное полное отсутствие верхней(их) конечности(ей)
- Q71.1. Врожденное отсутствие плеча и предплечья при наличии кисти
- Q71.2. Врожденное отсутствие предплечья и кисти
- Q71.3. Врожденное отсутствие кисти и пальца(ев)
- Q71.4. Продольное укорочение лучевой кости
- Q71.
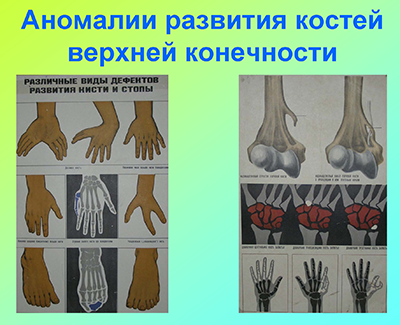 5. Продольное укорочение локтевой кости
5. Продольное укорочение локтевой кости - Q71.6. Клешнеобразная кисть
- Q71.8. Другие дефекты, укорачивающие верхнюю(ие) конечность(и)
- Q71.9. Дефект, укорачивающий верхнюю конечность, неуточненный
Q72. Дефекты, укорачивающие нижнюю конечность
- Q72.0. Врожденное полное отсутствие нижней(их) конечности(ей)
- Q72.1. Врожденное отсутствие бедра и голени при наличии стопы
- Q72.2. Врожденное отсутствие голени и стопы
- Q72.3. Врожденное отсутствие стопы и пальца(ев) стопы
- Q72.4. Продольное укорочение бедренной кости
- Q72.5. Продольное укорочение большеберцовой кости
- Q72.6. Продольное укорочение малоберцовой кости
- Q72.7. Врожденное расщепление стопы
- Q72.8. Другие дефекты, укорачивающие нижнюю(ие) конечность(и)
- Q72.9. Дефект, укорачивающий нижнюю конечность, неуточненный
Q73. Дефекты, укорачивающие конечность неуточненную
- Q73.0. Врожденное отсутствие конечности(ей) неуточненной(ых)
- Q73.
 1. Фокомелия конечности(ей) неуточненной(ых)
1. Фокомелия конечности(ей) неуточненной(ых) - Q73.8. Другие дефекты, укорачивающие конечность(и) неуточненную(ые)
Q74. Другие врожденные аномалии (пороки развития) конечности(ей)
- Q74.0. Другие врожденные аномалии верхней конечности(ей), включая плечевой пояс
- Q74.1. Врожденная аномалия коленного сустава
- Q74.2. Другие врожденные аномалии нижней(их) конечности(ей), включая тазовый пояс
- Q74.3. Врожденный множественный артрогрипоз
- Q74.8. Другие уточненные врожденные аномалии конечности(ей)
- Q74.9. Врожденная аномалия конечности(ей) неуточненная
Q75. Другие врожденные аномалии (пороки развития) костей черепа и лица
- Q75.0. Краниосиностоз
- Q75.1. Краниофациальный дизостоз
- Q75.2. Гипертелоризм
- Q75.3. Макроцефалия
- Q75.4. Челюстно-лицевой дистоз
- Q75.5. Окуломандибулярный дистоз
- Q75.8. Другие уточненные пороки развития костей черепа и лица
- Q75.
 9. Врожденная аномалия костей черепа и лица неуточненная
9. Врожденная аномалия костей черепа и лица неуточненная
Q76. Врожденные аномалии (пороки развития) позвоночника и костей грудной клетки
- Q76.0. Spina bifida occulta
- Q76.1. Синдром Клиппеля-Фейля
- Q76.2. Врожденный спондилолистез
- Q76.3. Врожденный сколиоз, вызванный пороком развития кости
- Q76.4. Другие врожденные аномалии позвоночника, не связанные со сколиозом
- Q76.5. Шейное ребро
- Q76.6. Другие врожденные аномалии ребер
- Q76.7. Врожденная аномалия грудины
- Q76.8. Другие врожденные аномалии костей грудной клетки
- Q76.9. Врожденная аномалия костей грудной клетки неуточненная
Q77. Отеоходродисплазия с дефектами роста трубчатых костей и позвоночника
- Q77.0. Ахондрогенезия
- Q77.1. Маленький рост, не совместимый с жизнью
- Q77.2. Синдром короткого ребра
- Q77.3. Точечная хондродисплазия
- Q77.4. Ахондроплазия
- Q77.5. Дистрофическая дисплазия
- Q77.
 6. Хондроэктодермальная дисплазия
6. Хондроэктодермальная дисплазия - Q77.7. Спондилоэпифизарная дисплазия
- Q77.8. Другая остеохондродисплазия с дефектами роста трубчатых костей и позвоночного столба
- Q77.9. Остеохондродисплазия с дефектами роста трубчатых костей и позвоночного столба неуточненная
Q78. Другие остеохондродисплазии
- Q78.0. Незавершенный остеогенез
- Q78.1. Полиостозная фиброзная дисплазия
- Q78.2. Остеопетроз
- Q78.3. Прогрессирующая диафизарная дисплазия
- Q78.4. Энхондроматоз
- Q78.5. Метафизарная дисплазия
- Q78.6. Множественные врожденные экзостозы
- Q78.8. Другие уточненные остеохондродисплазии
- Q78.9. Остеохондродисплазия неуточненная
Q79. Врожденные аномалии (пороки развития) костно-мышечной системы, не классифицированные в других рубриках
- Q79.0. Врожденная диафрагмальная грыжа
- Q79.1. Другие пороки развития диафрагмы
- Q79.2. Экзомфалоз
- Q79.3. Гастрошиз
- Q79.
 4. Синдром сливообразного живота
4. Синдром сливообразного живота - Q79.5. Другие врожденные аномалии брюшной стенки
- Q79.6. Синдром Элерса-Данло
- Q79.8. Другие пороки развития костно-мышечной системы
- Q79.9. Врожденный порок развития костно-мышечной системы неуточненный
Q80-Q89. Другие врожденные аномалии (пороки развития)
Q80. Врожденный ихтиоз
- Q80.0. Ихтиоз простой
- Q80.1. Ихтиоз, связанный с Х-хромосомой (Х-сцепленный ихтиоз)
- Q80.2. Пластинчатый (ламинарный) ихтиоз
- Q80.3. Врожденная буллезная ихтиозиформная эритродермия
- Q80.4. Ихтиоз плода («плод Арлекин»)
- Q80.8. Другой врожденный ихтиоз
- Q80.9. Врожденный ихтиоз неуточненный
Q81. Буллезный эпидермолиз
- Q81.0. Эпидермолиз буллезный простой
- Q81.1. Эпидермолиз буллезный летальный
- Q81.2. Эпидермолиз буллезный дистрофический
- Q81.8. Другой буллезный эпидермолиз
- Q81.9. Буллезный эпидермолиз неуточненный
Q82.
 Другие врожденные аномалии (пороки развития) кожи
Другие врожденные аномалии (пороки развития) кожи
- Q82.0. Наследственная лимфедема
- Q82.1. Ксеродерма пигментная
- Q82.2. Мастоцитоз
- Q82.3. Недержание пигмента (incontinentia pigmenti)
- Q82.4. Эктодермальная дисплазия (ангидротическая)
- Q82.5. Врожденный неопухолевый невус
- Q82.8. Другие уточненные врожденные аномалии кожи
- Q82.9. Врожденная аномалия развития кожи неуточненная
Q83. Врожденные аномалии (пороки развития) молочной железы
- Q83.0. Отсутствие молочной железы и соска
- Q83.1. Добавочная молочная железа
- Q83.2. Отсутствие соска
- Q83.3. Добавочный сосок
- Q83.8. Другие врожденные аномалии молочной железы
- Q83.9. Врожденная аномалия молочной железы неуточненная
Q84. Другие врожденные аномалии (пороки развития) наружных покровов
- Q84.0. Врожденная алопеция
- Q84.1. Врожденные морфологические нарушения волос, не классифицированные в других рубриках
- Q84.
 2. Другие врожденные аномалии волос
2. Другие врожденные аномалии волос - Q84.3. Анонихия
- Q84.4. Врожденная лейконихия
- Q84.5. Увеличенные и гипертрофированные ногти
- Q84.6. Другие врожденные аномалии ногтей
- Q84.8. Другие врожденные аномалии наружных покровов
- Q84.9. Порок развития наружных покровов неуточненный
Q85. Факоматозы, не классифицированные в других рубриках
- Q85.0. Нейрофиброматоз (незлокачественный)
- Q85.1. Туберозный склероз
- Q85.8. Другие факоматозы, не классифицированные в других рубриках
- Q85.9. Факоматоз неуточненный
Q86. Синдромы врожденных аномалий (пороков развития), обусловленные известными экзогенными факторами, не классифицированными в других рубриках
- Q86.0. Алкогольный синдром у плода (дизморфия)
- Q86.1. Синдром гидантоинового плода
- Q86.2. Дизморфия, вызванная варфарином
- Q86.8. Другие синдромы врожденных аномалий, обусловленных воздействием известных экзогенных факторов
Q87.
 Другие уточненные синдромы врожденных аномалий (пороков развития), затрагивающих несколько систем
Другие уточненные синдромы врожденных аномалий (пороков развития), затрагивающих несколько систем
- Q87.0. Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
- Q87.1. Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью
- Q87.2. Синдромы врожденных аномалий, вовлекающие преимущественно конечности
- Q87.3. Синдромы врожденных аномалий, проявляющиеся избыточным ростом (гигантизмом) на ранних этапах развития
- Q87.4. Синдром Марфана
- Q87.5. Другие синдромы врожденных аномалий с другими изменениями скелета
- Q87.8. Другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках
Q89. Другие врожденные аномалии (пороки развития), не классифицированные в других рубриках
- Q89.0. Врожденные аномалии селезенки
- Q89.1. Пороки развития надпочечника
- Q89.2. Врожденные аномалии других эндокринных желез
- Q89.3. Situs inversus
- Q89.
 4. Сросшаяся двойня
4. Сросшаяся двойня - Q89.7. Множественные врожденные аномалии, не классифицированные в других рубриках
- Q89.8. Другие уточненные врожденные аномалии
- Q89.9. Врожденная аномалия неуточненная
Q90-Q99. Хромосомные аномалии, не классифицированные в других рубриках
Q90. Синдром Дауна
- Q90.0. Трисомия 21, мейотическое нерасхождение
- Q90.1. Трисомия 21, мозаицизм (митотическое нерасхождение)
- Q90.2. Трисомия 21, транслокация
- Q90.9. Синдром Дауна неуточненный
Q91. Синдром Эдвардса и синдром Патау
- Q91.0. Трисомия 18, мейотическое нерасхождение
- Q91.1. Трисомия 18, мозаицизм (митотическое нерасхождение)
- Q91.2. Трисомия 18, транслокация
- Q91.3. Синдром Эдвардса неуточненный
- Q91.4. Трисомия 13, мейотическое нерасхождение
- Q91.5. Трисомия 13, мозаицизм (митотическое нерасхождение)
- Q91.6. Трисомия 13, транслокация
- Q91.7. Синдром Патау неуточненный
Q92.
 Другие трисомии и частичные трисомии аутосом, не классифицированные в других рубриках
Другие трисомии и частичные трисомии аутосом, не классифицированные в других рубриках
- Q92.0. Полная хромосомная трисомия, мейотическое нерасхождение
- Q92.1. Полная хромосомная трисомия, мозаицизм (митотическое нерасхождение)
- Q92.2. Большая частичная трисомия
- Q92.3. Малая частичная трисомия
- Q92.4. Удвоения, наблюдаемые только в прометафазе
- Q92.5. Удвоения с другим комплексом перестроек
- Q92.6. Особо отмеченные хромосомы
- Q92.7. Триплоидия и полиплоидия
- Q92.8. Другие уточненные трисомии и частичные трисомии аутосом
- Q92.9. Трисомии и частичные трисомии аутосом неуточненные
Q93. Моносомии и утраты части аутосом, не классифицированные в других рубриках
- Q93.0. Полная хромосомная моносомия, мейотическое нерасхождение
- Q93.1. Полная хромосомная моносомия, мозаицизм (митотическое нерасхождение)
- Q93.2. Хромосомное смещение с закруглением или смещением центра
- Q93.
 3. Делеция короткого плеча хромосомы 4
3. Делеция короткого плеча хромосомы 4 - Q93.4. Делеция короткого плеча хромосомы 5
- Q93.5. Другие делеции части хромосомы
- Q93.6. Делеции, наблюдаемые только в прометафазе
- Q93.7. Делеции с другим комплексом перестроек
- Q93.8. Другие делеции из аутосом
- Q93.9. Делеция из аутосом неуточненная
Q95. Сбалансированные перестройки и структурные маркеры, не классифицированные в других рубриках
- Q95.0. Сбалансированные транслокации и инсерции у нормального индивида
- Q95.1. Хромосомные инверсии у нормального индивида
- Q95.2. Сбалансированные аутосомные перестройки у нормального индивида
- Q95.3. Сбалансированные половые/аутосомные перестройки у нормального индивида
- Q95.4. Индивиды с обозначенным гетерохроматином
- Q95.5. Индивиды с ломким участком аутосом
- Q95.8. Другие сбалансированные перестройки и структурные маркеры
- Q95.9. Сбалансированные перестройки и структурные маркеры неуточненные
Q96.
 Синдром Тернера
Синдром Тернера
- Q96.0. Кариотип 45,Х
- Q96.1. Кариотип 46,Х iso (Xq)
- Q96.2. Кариотип 46,Х с аномальной половой хромосомой, за исключением iso (Xq)
- Q96.3. Мозациизм, 45,Х/46,ХХ или ХУ
- Q96.4. Мозациизм, 45,Х/другая клеточная линия (линии) с аномальной половой хромосомой
- Q96.8. Другие варианты синдрома Тернера
- Q96.9. Синдром Тернера неуточненный
Q97. Другие аномалии половых хромосом, женский фенотип, не классифицированные в других рубриках
- Q97.0. Кариотип 47,ХХХ
- Q97.1. Женщина с более чем тремя Х-хромосомами
- Q97.2. Мозациизм, цепочки с различным числом Х-хромосом
- Q97.3. Женщина с 46,ХУ-кариотипом
- Q97.8. Другие уточненные аномальные половые хромосомы, женский фенотип
- Q97.9. Аномалия половых хромосом, женский фенотип, неуточненная
Q98. Другие аномалии половых хромосом, мужской фенотип, не классифицированные в других рубриках
- Q98.0. Синдром Клайнфелтера, кариотип 47,ХХУ
- Q98.
 1. Синдром Клайнфелтера, мужчина с более чем двумя Х-хромосомами
1. Синдром Клайнфелтера, мужчина с более чем двумя Х-хромосомами - Q98.2. Синдром Клайнфелтера, мужчина с 46,ХХ-кариотипом
- Q98.3. Другой мужчина с 46,ХХ-кариотипом
- Q98.4. Синдром Клайнфелтера неуточненый
- Q98.5. Кариотип 47,ХУУ
- Q98.6. Мужчина со структурно измененными половыми хромосомами
- Q98.7. Мужчина с мозаичными половыми хромосомами
- Q98.8. Другие уточненные аномалии половых хромосом, мужской фенотип
- Q98.9. Аномалия половых хромосом, мужской фенотип, неуточненная
Q99. Другие аномалии хромосом, не классифицированные в других рубриках
- Q99.0. Мозаик (химера) 46,ХХ/46,ХУ
- Q99.1. 46,ХХ истинный гермафродит
- Q99.2. Ломкая Х-хромосома
- Q99.8. Другие уточненные хромосомные аномалии
- Q99.9. Хромосомная аномалия неуточненная
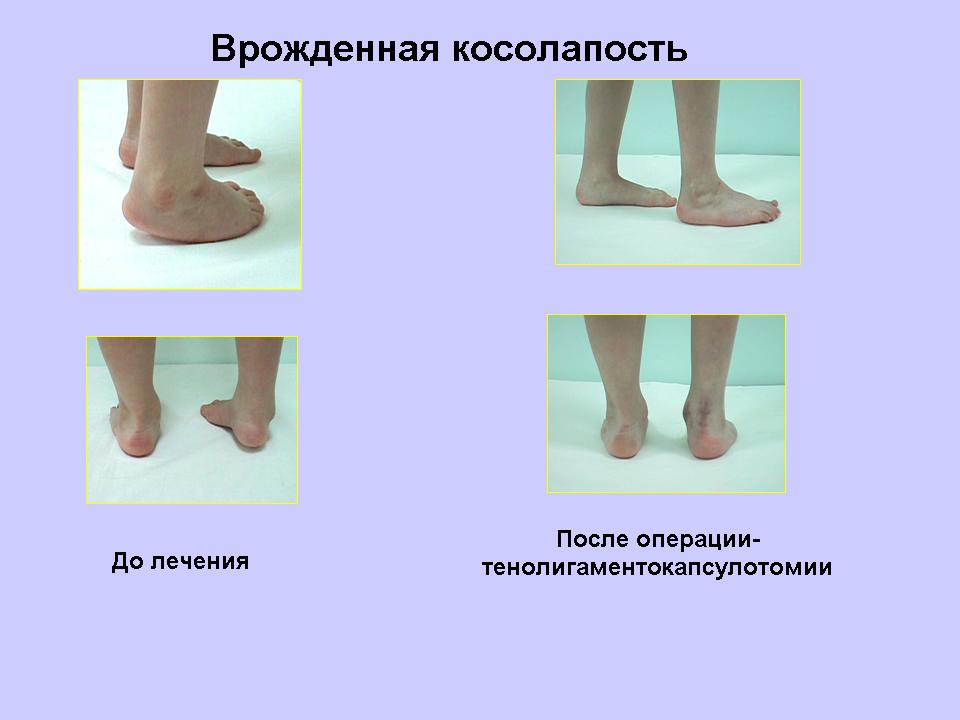
Деформации стоп и их лечение — (клиники Di Центр)
Человеческая стопа несёт на себе колоссальную нагрузку, позволяя человеку свободно передвигаться в пространстве. Колебания массы тела всегда заканчиваются изменениями в области свода.
Колебания массы тела всегда заканчиваются изменениями в области свода.
Любые деформации характеризуются стойким изменением натурального вида. Подобные изменения могут коснуться длины отдельных костей, а также прочности сухожилий и связочного аппарата. Когда у человека происходят деформирующие изменения конечностей, у него нарушается походка, появляется дискомфорт и болезненность при физической нагрузке.
Причиной таких изменений является неправильное распределение человеческого веса по всей площади стоп. Деформирующие изменения возникают с одинаковой частотой у представителей мужского и женского пола, независимо от возраста. В группу риска попадают люди, страдающие хроническими заболеваниями костно-суставного аппарата, спортсмены, а также лица, чья трудовая деятельность связана с избыточной нагрузкой на ноги.
Причины деформации стоп
Самыми распространенными причинами деформации можно назвать:
-
травмы; -
заболевания соединительной ткани; -
наследственная предрасположенность; -
длительное нахождение в условиях низкой температуры; -
врождённые пороки развития костно-суставного аппарата.
Даже незначительная деформация области стопы может послужить причиной развития таких болезней, как артроз и остеохондроз. К тому же, у людей, имеющих деформации, наблюдается изменение осанки, вплоть до сколиоза.
Виды деформации стоп
В зависимости от места локализации патологических изменений, принято выделять такие виды деформаций стоп:
-
Молоткообразное искривление фаланг. Данная патология чаще всего затрагивает II и III пальцы, которые приобретают форму молоточков. На фоне искривления возникают такие осложнения как, мозоли в отдельных участках стопы, а также остеоартроз.
-
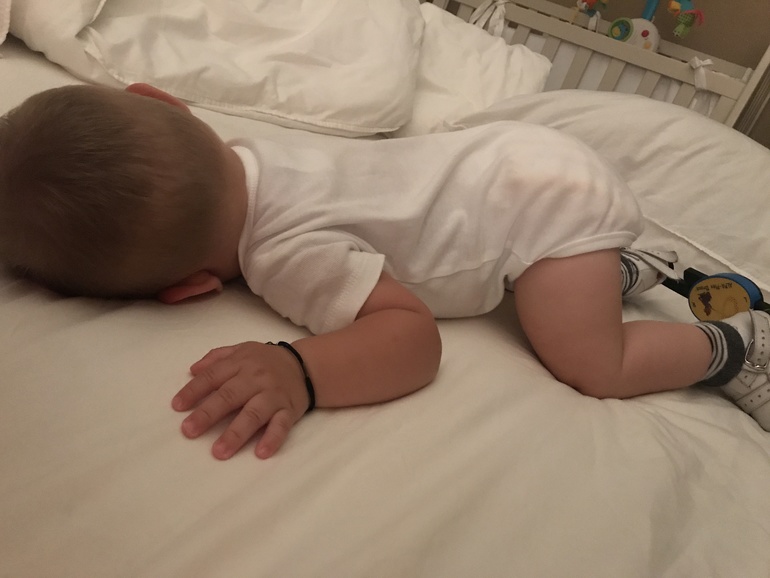
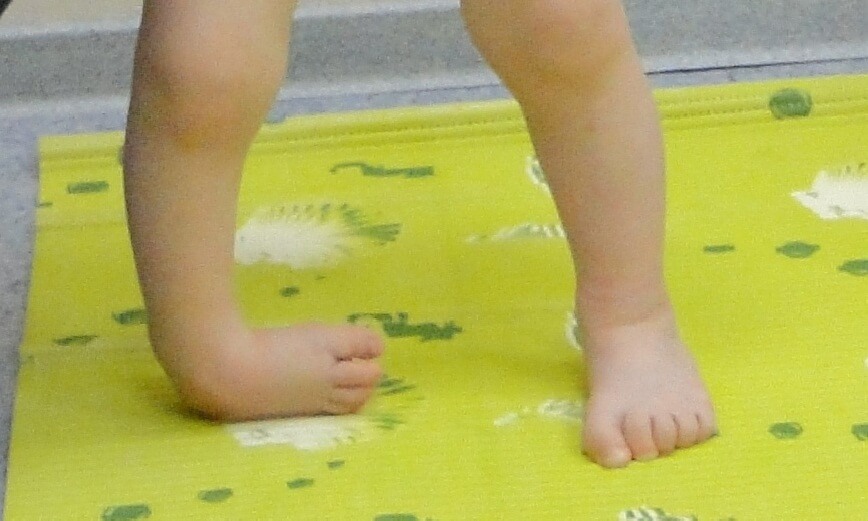
Косолапость. Этот вид деформирующих изменений наиболее часто встречается в повседневной практике. Чаще всего, заболевание диагностируется у детей в дошкольном и младшем школьном возрасте. Для косолапости характерно уменьшение длины стопы, и её супинация. Причина косолапости — подвывих голеностопного сустава. Эта болезнь может быть как приобретённой, так и врождённой. Приобретённая форма болезни формируется в результате травм, парезов и параличей. В результате неправильного распределения нагрузки, у человека формируются мозоли на тех участках стопы, которые выступают опорой при ходьбе.
Для косолапости характерно уменьшение длины стопы, и её супинация. Причина косолапости — подвывих голеностопного сустава. Эта болезнь может быть как приобретённой, так и врождённой. Приобретённая форма болезни формируется в результате травм, парезов и параличей. В результате неправильного распределения нагрузки, у человека формируются мозоли на тех участках стопы, которые выступают опорой при ходьбе.
-
Сводчатая ступня. Это состояние выражается в виде усиления искривления продольной части стопы. При тяжёлом течении патологии у человека не соприкасается середина стопы и поверхность, на которой он стоит. -
Плоскостопие. Неправильное распределение веса человеческого тела очень часто провоцирует уплощение поперечной или продольной области свода. Если у человека сформировалось продольное плоскостопие, то нагрузка распространяется на всю стопу, а не вдоль её наружного края (в норме). Для поперечного плоскостопия характерно увеличение промежутков между головками плюсневых костей, и расширение передних отделов стопы.
Для поперечного плоскостопия характерно увеличение промежутков между головками плюсневых костей, и расширение передних отделов стопы.
-
Пяточная стопа. Для этого заболевания характерно постоянное сгибание тыльной части стопы. При тяжёлом течении патологии, у человека может наблюдаться соприкосновение тыльной части стопы и передней поверхности голени.
-
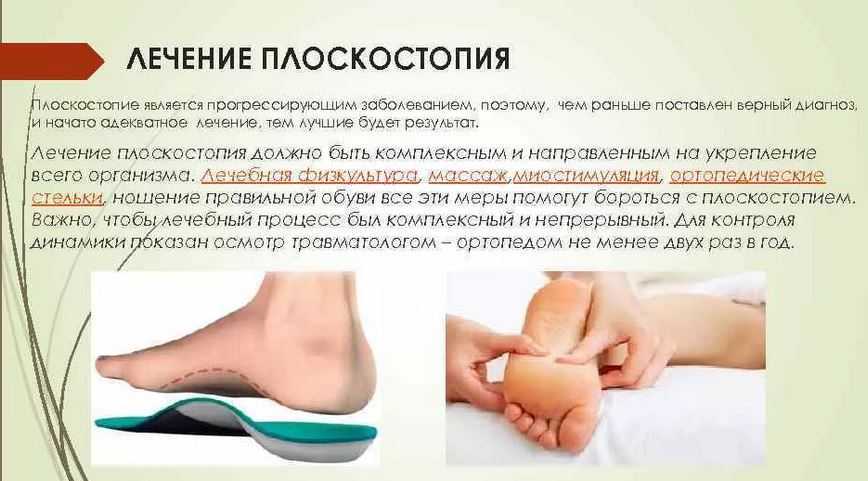
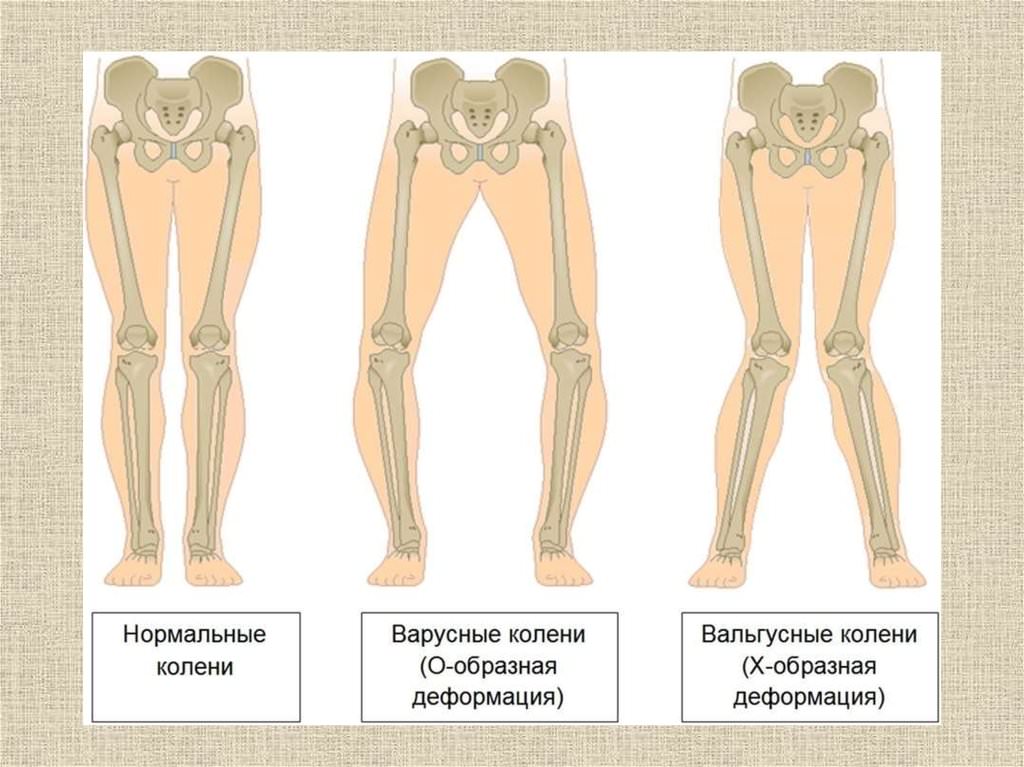
Вальгусные и варусные стопы. Такая патология встречается чаще всего у детей, при вальгусной стопе ноги принимают х-образную форму, а пир варусной — о-образную. -
Вальгусная деформация большого пальца ноги сопровождается образованием косточки у основания большого пальца. Патология встречается чаще всего у женщин, которые постоянно носят обувь на высоких каблуках.
Есть еще врожденный вид деформаций. Эта разновидность деформаций встречается не так часто, однако нельзя исключать её полностью. Ярким представителем врождённых патологий является эквинусная деформация стопы и вертикальная деформация области таранной кости. Это заболевание обусловлено врождёнными пороками развития фиброзной (соединительной) ткани. В период возрастного роста опорно-двигательного аппарата мышцы не успевают за темпами роста отдельных костей, в результате чего происходит их смещение за пределы анатомической нормы.
Эта разновидность деформаций встречается не так часто, однако нельзя исключать её полностью. Ярким представителем врождённых патологий является эквинусная деформация стопы и вертикальная деформация области таранной кости. Это заболевание обусловлено врождёнными пороками развития фиброзной (соединительной) ткани. В период возрастного роста опорно-двигательного аппарата мышцы не успевают за темпами роста отдельных костей, в результате чего происходит их смещение за пределы анатомической нормы.
Признаки
Для каждой разновидности деформации характерна своя клиническая картина. В качестве общих признаков нарушений формы стопы, можно выделить:
Для косолапости характерна невозможность поворота ноги вовнутрь и поворота носка. При разных видах плоскостопия у человека наблюдается тяжёлая походка. С целью снижения болевого синдрома человек старается опираться на ребро ноги во время ходьбы. Изменение формы ступней при молоткообразном искривлении и вальгусной деформации становиться причиной невозможности ношения тесной обуви, а также обуви на каблуках.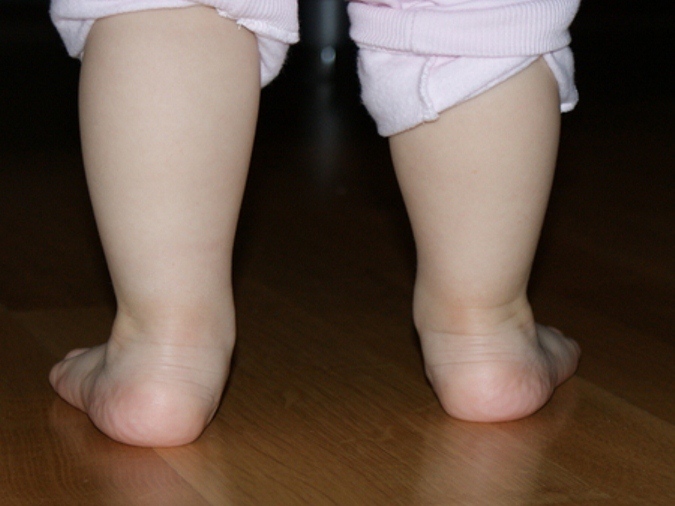
Диагностика
При постановке диагноза медицинский специалист учитывает жалобы пациента, а также данные клинического осмотра. Обязательными критериями является положение стопы, объём движений в голеностопных и плюсневых суставах, а также внешний вид ног. Для получения достоверного диагноза используются такие методы:
-
Рентгенографическое исследование стопы и голеностопного сустава. Данный вид исследования позволяет обнаружить место локализации деформирующих изменений. -
Электромиография. Методика измерения биоэлектрической активности мышц широко используется при диагностике конской стопы. На основании данных исследования можно определить сократительный потенциал мышц стопы. -
Магнитно-резонансная и компьютерная томография. Эти методы применяются при малой информативности других исследований.
Лечение деформации стоп
Каждый вид деформации нуждается в индивидуальном подходе. Для коррекции некоторых заболеваний бывает достаточно консервативного лечения, а в других клинических случаях пациенту рекомендовано оперативное вмешательство.
-
Косолапость
При врождённой форме заболевания коррекция стопы начинается с первых дней жизни ребёнка. Детский ортопед производит ручное выведение ножки грудничка в правильное анатомическое положение, после чего её фиксируют гипсовой повязкой. Со временем гипс меняют на шину, которая накладывается перед сном. Когда малышу исполняется 3 года, ему назначают лечебную физкультуру и массаж.
Консервативное лечение приобретённой косолапости заключается в использовании специальных вкладышей и ортопедической обуви. В качестве дополнительных методик используется физиотерапия и лечебная физкультура.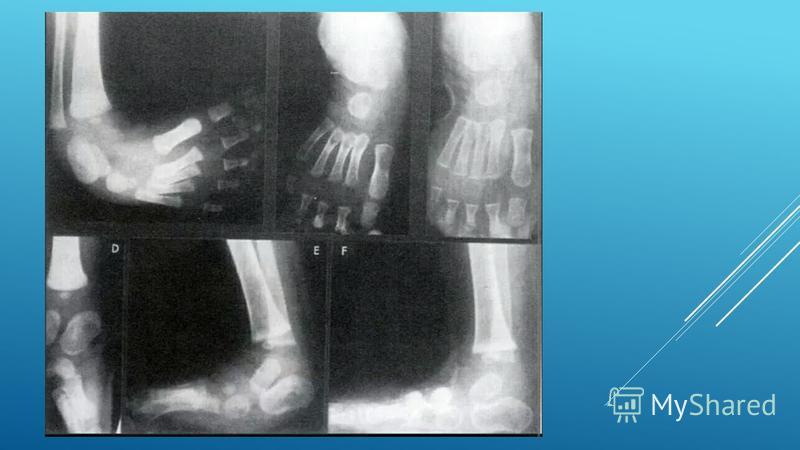 При недостаточной эффективности данного лечения пациентам выполняется артродез.
При недостаточной эффективности данного лечения пациентам выполняется артродез.
-
Плоскостопие
Лечение всех видов плоскостопия осуществляется с использованием консервативных методов. Таким пациентам рекомендуют ношение ортопедических стелек, коррекция гипсовыми повязками, лечебную физкультуру, массаж и аппаратную физиотерапию.
-
Конская стопа
Для ликвидации этого вида деформации используются фиксирующие шины и редрессирующие повязки, корректоры для стопы и пальцев ног, ортопедическая обувь, бандажи, специализированные тяги и пяточные шины. Если консервативная терапия не дала положительного результата, то пациентам назначают артродез голеностопного сустава.
-
Пяточная стопа
Врождённая форма заболевания поддаётся консервативному лечению посредством редрессирующих гипсовых повязок и фиксирующих шин. Если деформация приобретенная, то исправить её сможет только хирургическое вмешательство.
Если деформация приобретенная, то исправить её сможет только хирургическое вмешательство.
-
Вальгусная деформация I пальца стопы
Для устранения шишки на ноге используются физиотерапевтические виды лечения, лечебная гимнастика, массаж, противовоспалительные мази, фиксирующие бандажи и шины. Если заболевание прогрессирует, то человеку назначают оперативное лечение.
Хирургическое лечение вальгусной деформации может осуществляться посредством таких методик: остеотомия проксимальной фаланги; удаление (экзостозэктомия) костного новообразования; рассечение мышцы, которая отводит поврежденный палец; остеотомия плюсневой кости.
Для того чтобы скорректировать форму пальцев, используются такие виды приспособлений, как ортопедические лонгеты, носки, накладки, вкладыши и колпачки. Также, пациентам рекомендован лечебный массаж, гимнастика, физиотерапия. При запущенном течении, молоткообразная деформация пальцев стопы корригируется при помощи хирургической операции. Этот вид лечения может быть как радикальным, так и симптоматическим.
Этот вид лечения может быть как радикальным, так и симптоматическим.
При возникновении деформирующих изменений, человеку может потребоваться дополнительная консультация врача-невролога. На протяжении всего периода терапии, рекомендован постоянный врачебный контроль динамики болезни.
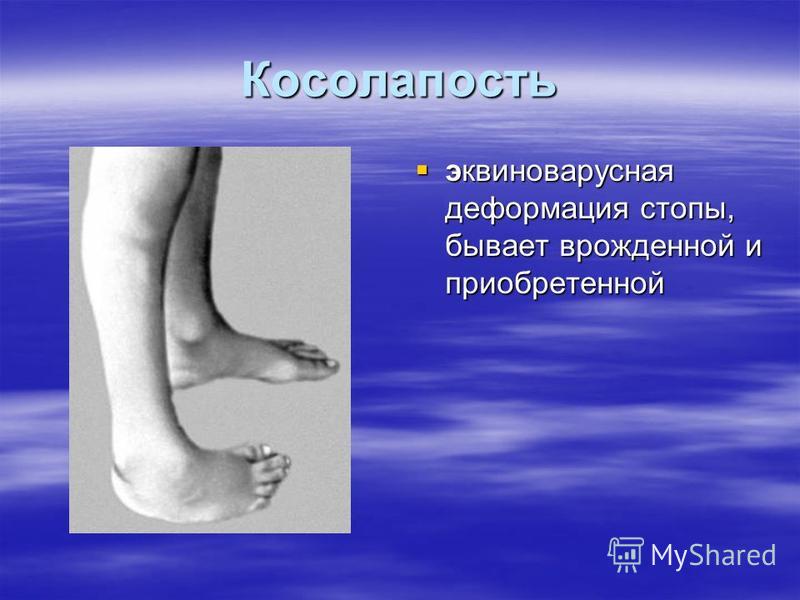
Эквиноварусная косолапость, что это такое, лечение
Обзор
Что такое косолапость?
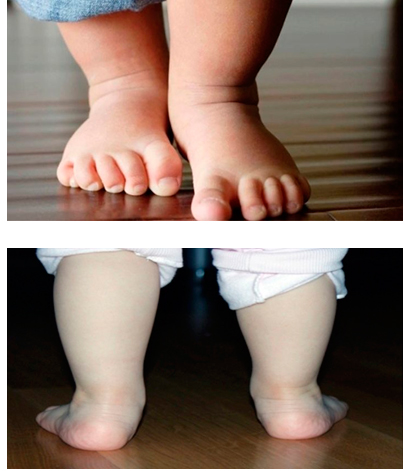
Косолапость, также называемая эквиноварусной косолапостью, представляет собой врожденный дефект, поражающий стопу и голеностопный сустав. Это врожденное заболевание, что означает, что ребенок рождается с ним. Стопа или стопы поворачиваются внутрь. Когда вы смотрите на стопу, нижняя часть стопы часто смотрит в сторону или даже вверх.
Косолапость возникает из-за проблем с сухожилиями, тканями, соединяющими мышцы с костями. Сухожилия в ноге и стопе ребенка короче и туже, чем должны быть. Это приводит к искривлению стопы.
Обширная хирургия раньше была основным методом лечения косолапости. Но сегодня поставщики медицинских услуг обычно используют комбинацию нехирургических методов и незначительной процедуры.
Но сегодня поставщики медицинских услуг обычно используют комбинацию нехирургических методов и незначительной процедуры.
Какие бывают виды косолапости?
Существует два типа косолапости:
- Изолированная или идиопатическая косолапость — наиболее распространенный тип. Если у вашего ребенка косолапость без каких-либо других медицинских проблем, это называется изолированной косолапостью. Идиопатический означает, что причина косолапости неизвестна.
- Неизолированная косолапость возникает наряду с другими проблемами со здоровьем. Эти состояния включают артрогрипоз (проблема с суставами) и расщепление позвоночника (заболевание нервной трубки). Дефекты нервной трубки — это проблемы головного, позвоночника и спинного мозга.
Кто подвержен риску врожденной косолапости?
Вероятность развития косолапости у мальчиков в два раза выше, чем у девочек. Наличие косолапости в семейном анамнезе также подвергает ребенка более высокому риску.
Младенцы также подвергаются повышенному риску, если у них есть:
- Другие врожденные дефекты, такие как расщелина позвоночника или церебральный паралич.
- Генетическое заболевание, такое как трисомия 18 (синдром Эдвардса).
Женщина может подвергаться повышенному риску рождения ребенка с косолапостью, если она:
- Во время беременности имела маловодие. Это проблема нехватки околоплодных вод, жидкости, которая окружает ребенка.
- Перенесла инфекцию Зика во время беременности, что может привести к врожденным дефектам и другим проблемам.
- Курила, употребляла алкоголь или наркотики во время беременности.
Врожденная косолапость поражает одну или обе стопы?
Около половины детей с косолапостью имеют проблемы с обеими стопами.
Как косолапость влияет на моего ребенка?
Косолапость не причиняет ребенку боли. Многие младенцы даже не замечают этого в течение первых нескольких месяцев жизни. Но косолапость будет мешать стоять и ходить. Это не пройдет само по себе. Младенцы с косолапостью нуждаются в лечении, чтобы устранить проблему, прежде чем они достигнут возраста ходьбы.
Но косолапость будет мешать стоять и ходить. Это не пройдет само по себе. Младенцы с косолапостью нуждаются в лечении, чтобы устранить проблему, прежде чем они достигнут возраста ходьбы.
Невылеченная косолапость может привести к:
- Проблемы с ходьбой. Дети с косолапостью часто ходят необычным образом. Как правило, люди ходят на ступнях и подошвах ног. Ребенок с косолапостью может ходить на боках и на верхней части стопы.
- Инфекции стопы.
- Проблемы со стопами, включая мозоли. Мозоль представляет собой толстый слой кожи, который часто развивается на подошве стопы.
- Артрит, заболевание суставов, вызывающее боль, скованность и отек.
Насколько распространена косолапость?
Косолапость — одна из самых распространенных проблем, с которыми рождаются дети. Примерно 1 из 1000 детей рождается с косолапостью.
Симптомы и причины
Что вызывает врожденную косолапость?
Исследователи не знают точную причину косолапости.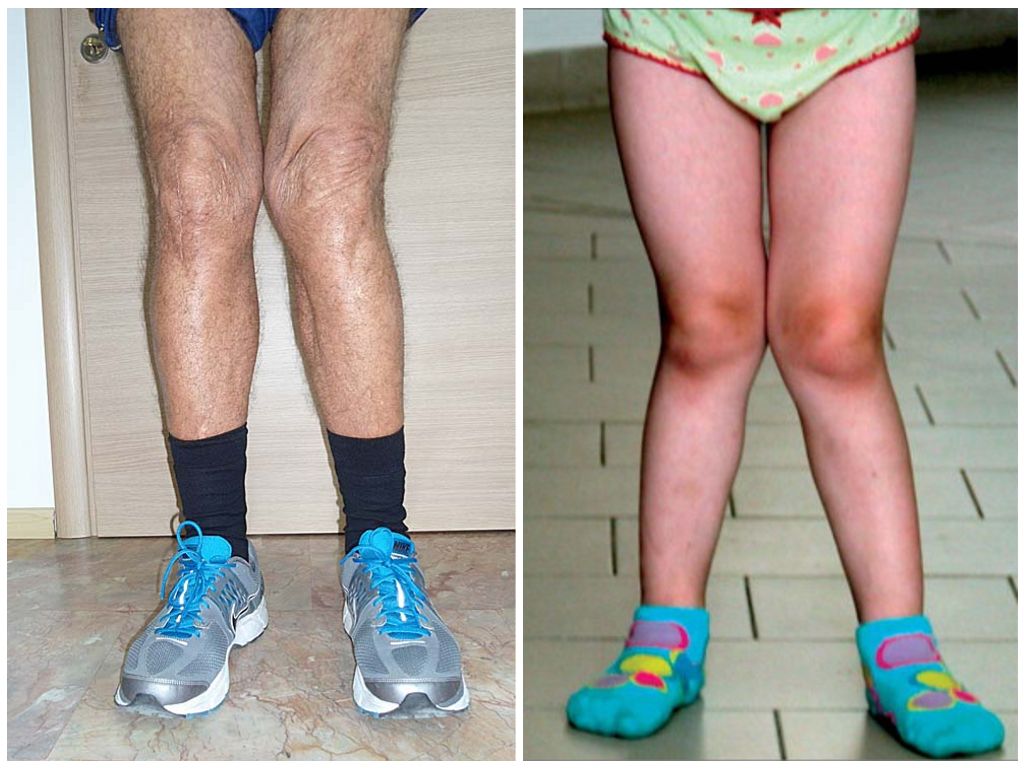 Скорее всего, это сочетание генетики и окружающей среды:
Скорее всего, это сочетание генетики и окружающей среды:
- Генетика: Гены говорят телу, как выглядеть, расти и функционировать. Проблема с одним или несколькими генами (которые передаются от родителей к детям) может привести к косолапости.
- Окружающая среда: Употребление наркотиков и курение во время беременности могут повышать риск рождения ребенка с врожденными дефектами, такими как косолапость.
Каковы симптомы косолапости?
Наиболее распространенным признаком косолапости является поворот одной или обеих стоп внутрь. Стопа обращена к противоположной ноге.
Вы также можете заметить, что стопа имеет:
- форму почки.
- Глубокая складка на внутренней средней части стопы.
- Свод выше нормы (так называемая полая деформация стопы).
Другие проблемы, которые вы можете заметить:
- Уменьшение икроножной мышцы в пораженной ноге.
- Укороченная лапка.

- Тугоподвижность голеностопного сустава.
- Отсутствие полной амплитуды движений в стопе.
Диагностика и тесты
Когда и как диагностируется косолапость?
Медицинский работник часто замечает косолапость во время УЗИ во время беременности. Пренатальное УЗИ показывает изображения вашего ребенка в матке (матке). Если ваш лечащий врач диагностирует косолапость во время беременности, вы можете начать планировать лечение, которое потребуется вашему ребенку после рождения.
В других случаях врач может диагностировать косолапость уже после рождения ребенка. Обычно они замечают это во время одного из первых медицинских осмотров ребенка. В некоторых случаях ваш врач может порекомендовать рентген для подтверждения диагноза.
Управление и лечение
Когда следует начинать лечение косолапости?
Медицинские работники рекомендуют лечить косолапость как можно скорее. Раннее лечение помогает ребенку избежать проблем в дальнейшем. Лучше всего начинать лечение в первые две недели жизни ребенка.
Лучше всего начинать лечение в первые две недели жизни ребенка.
Кто лечит косолапость?
Для лечения косолапости вашему ребенку, скорее всего, понадобится команда врачей, в том числе:
- Детский ортопед, специализирующийся на проблемах костей и суставов у детей.
- Хирург-ортопед, специализирующийся на хирургии костей и суставов.
- Физиотерапевт, чтобы помочь ребенку набраться сил и двигать стопой.
Как лечить косолапость?
Существует несколько методов лечения косолапости. Ваша команда по уходу обсудит с вами варианты и решит, какой из них лучше всего подходит для вашего ребенка. Процедуры включают:
- Метод Понсети, , который растягивает и гипсует ногу, чтобы исправить искривление.
- Французский метод, , который растягивает и накладывает шину на ногу для исправления искривления.
- Распорка, с использованием специальной обуви для удержания стопы под правильным углом.
- Хирургия, , который может быть вариантом, если другие методы не работают.
Что такое метод Понсети для лечения косолапости?
Метод Понсети является наиболее популярным методом лечения, включающим серийное литье. Он длится около двух-трех месяцев. Ваша команда по уходу начнет эту терапию в течение первых двух недель после рождения.
Этот метод выполняет хирург-ортопед. Они будут:
- Вытягивать стопу в правильное положение.
- Наложите на стопу гипсовую повязку, которая начинается от пальцев ног и доходит до верхней части бедра.
- Повторяйте этот процесс каждые четыре-семь дней с новым гипсом. Каждый раз хирург немного приближает стопу к правильному положению.
Перед окончательной повязкой хирург обычно выполняет тенотомию ахиллова сухожилия. Они:
- Быстро перережьте пяточную связку (ахиллово сухожилие). Это сухожилие соединяет пятку с икроножными мышцами. Вырез небольшой и не требует швов.
- Наложите новую повязку, когда сухожилие заживет, что занимает около трех недель.
Целью этой операции является увеличение сухожилия до нормальной длины. Когда сняли последний гипс, сухожилие достигло нормальной длины. По мере выздоровления вашего ребенка ему может понадобиться:
- Делать упражнения на растяжку, чтобы стопы оставались в правильном положении.
- Носите специальную обувь или ортез в течение нескольких лет.
Что представляет собой французский метод лечения косолапости?
Этот метод похож на метод Понсети, но вместо гипсовой повязки используется шинирование и тейпирование. Шина — это устройство, которое поддерживает и защищает кости.
Это лечение проводит физиотерапевт. Они начнут лечение вскоре после рождения. Это лечение нужно делать каждый день, а не один раз в неделю. Но вам не нужно каждый раз возвращаться к физиотерапевту. Физиотерапевт осматривает вашего ребенка несколько раз в неделю и учит вас, как делать шинирование и тейпирование в домашних условиях.
Как исправить косолапость по французской методике:
- Вытяните стопу ребенка в правильное положение.
- Закрепите стопу с помощью ленты и шин.
- Повторяйте этот процесс каждый день в течение двух месяцев.
- Повторяйте процедуру реже, пока ребенку не исполнится 3 месяца. (Физический терапевт скажет вам, как часто это делать).
Младенцам, перенесшим операцию по французскому методу, также часто требуется тенотомия ахиллова сухожилия.
Через три месяца вы, вероятно, заметите улучшение стопы вашего ребенка. Чтобы сохранить правильное положение стопы и предотвратить повторное появление косолапости, родителям часто приходится продолжать лечение до тех пор, пока их ребенку не исполнится 2 или 3 года.
Как лечат косолапость с помощью корсетов?
Ваша команда по уходу может порекомендовать ношение корсета после того, как ваш ребенок закончит использовать метод Понсети или французский метод. Даже если эти методы лечения сработали, стопа может вернуться в неправильное положение. Корсет удерживает ногу под правильным углом, поэтому она не смещается. Подпорка обычно представляет собой пару обуви с металлическим стержнем, соединяющим их. Бандаж часто называют «сапоги и штанга». Важно:
Корсет удерживает ногу под правильным углом, поэтому она не смещается. Подпорка обычно представляет собой пару обуви с металлическим стержнем, соединяющим их. Бандаж часто называют «сапоги и штанга». Важно:
- Носить корсет каждый день в течение трех месяцев, затем только ночью или во время сна в течение четырех лет.
- Внимательно следуйте инструкциям. Если ребенок не наденет брекеты в положенное время, стопа может снова вернуться в положение косолапости.
Существует несколько типов брекетов. Ваш врач обсудит с вами варианты, чтобы вы могли подобрать правильный корсет для своего ребенка.
Как хирургия лечит косолапость?
Иногда у ребенка тяжелая форма косолапости. Или вы пробовали нехирургические методы, но они не сработали. Хирургия может исправить проблему. Лучше всего, если вашему ребенку сделают операцию до того, как он начнет ходить. Во время процедуры хирург:
- Удлиняет пяточный шнур и устраняет другие проблемы со стопой или стопами.
- Вставляет штифты в опору для исправления положения.
- Накладывает гипс на ногу после операции.
Через несколько недель после операции хирург:
- Снимает гипс и штифты.
- Надевает новую повязку на ногу ребенка, которую ваш ребенок носит еще около четырех недель.
- Удаляет окончательный слепок.
Еще есть шанс, что стопа вернется в положение косолапости. Ваш врач может порекомендовать ортезы или специальную обувь, чтобы стопа оставалась в правильном положении.
Каковы риски хирургического лечения косолапости?
Риски операции по поводу врожденной косолапости включают:
- Повреждение нерва.
- Инфекция.
- Кровотечение.
- Жесткость.
Профилактика
Как предотвратить косолапость у ребенка?
Хорошее медицинское обслуживание до и во время беременности дает вашему ребенку наилучшие шансы на здоровое начало жизни. Еще до того, как вы забеременеете, вы можете подумать, подходит ли вам обследование перед зачатием. Во время этого визита медицинский работник удостоверится, что вы в максимально возможной степени здоровы, когда забеременеете.
Во время этого визита медицинский работник удостоверится, что вы в максимально возможной степени здоровы, когда забеременеете.
Если вы подвержены высокому риску рождения ребенка с косолапостью или другими врожденными дефектами, поговорите с консультантом-генетиком. Генетический консультант является экспертом по врожденным дефектам и генетическим заболеваниям. И проверьтесь на инфекции, такие как вирус Зика. Лечение инфекций до того, как вы забеременеете, увеличивает шансы на здоровую беременность и ребенка.
Если вы беременны:
- Обязательно посещайте все дородовые осмотры.
- Защита от вируса Зика.
- Не курите, не употребляйте запрещенные наркотики и не употребляйте алкоголь.
Перспективы/прогноз
Каковы перспективы для детей с косолапостью?
Косолапость не проходит сама по себе. Раннее лечение имеет важное значение для положительного результата. Младенцы, которые рано начинают лечение, имеют хорошие результаты. Они могут носить обычную обувь, ходить, бегать и играть без боли. Они даже могут заниматься спортом.
Они могут носить обычную обувь, ходить, бегать и играть без боли. Они даже могут заниматься спортом.
Если поражена только одна стопа, вы можете заметить, что:
- Пораженная стопа меньше по размеру и менее подвижна, чем здоровая стопа.
- Икроножные мышцы в ноге при косолапости могут быть меньше.
- Ваш ребенок может устать или жаловаться на боли в ногах раньше, чем дети без косолапости.
- Пораженная нога может быть немного короче. Но обычно это не вызывает серьезных проблем.
Если у вашего ребенка наряду с косолапостью имеется другое заболевание, прогноз может зависеть от лечения другого заболевания.
Может ли косолапость вернуться?
Косолапость может вернуться. Это более вероятно, если график лечения не соблюдался точно. Если стопа возвращается в положение косолапости, обратитесь к врачу. Они могут посоветовать вам дальнейшие действия. Возможно, вам придется повторить некоторые этапы плана лечения.
Жить с
Как я могу помочь моему ребенку с корсетом?
Регулярное ношение корсета повышает шансы вашего ребенка на успех. Но детям может быть сложно носить корсет столько часов в день. Эти советы могут помочь родителям облегчить процесс ношения корсета:
Но детям может быть сложно носить корсет столько часов в день. Эти советы могут помочь родителям облегчить процесс ношения корсета:
- Развлекайтесь: Играйте с ребенком, когда он носит корсет. Выполняйте легкие упражнения и игры с ногами. Используйте штангу, чтобы помочь согнуть и выпрямить колени.
- Сделайте это привычным: По прошествии первых трех месяцев вашему ребенку будет нужен бандаж только в ночное время и во время дневного сна. Сделайте корсет частью их режима сна. Они поймут, что спать означает носить корсет.
- Добавьте прокладку: Мягкая прокладка на металлической планке делает бандаж более удобным для вашего ребенка и для вас. Он также имеет преимущество защиты мебели и бытовой техники в вашем доме.
- Избегайте лосьона: Кремы или лосьоны могут усугубить проблемы с кожей. Это нормально, если стопа вашего ребенка немного покраснела. Но волдыри могут означать, что пятка выскальзывает из скобы.
 Обязательно закрепите обувь, чтобы нога не соскальзывала. И часто проверяйте ногу вашего ребенка, чтобы убедиться, что волдыри не образуются.
Обязательно закрепите обувь, чтобы нога не соскальзывала. И часто проверяйте ногу вашего ребенка, чтобы убедиться, что волдыри не образуются. - Предотвращение соскальзывания: Иногда стопа продолжает соскальзывать с бандажа. Убедитесь, что ремешок туго затянут. Двойные носки также могут помочь надежно удерживать обувь на ноге. Физиотерапевт может порекомендовать другие шаги, чтобы убедиться, что обувь плотно прилегает к ноге.
Что еще я должен спросить у своего лечащего врача о косолапости?
Попросите у своего врача направление к хирургу-ортопеду, специализирующемуся на методе Понсети. Это лечение требует высокого уровня мастерства и опыта. Если ваша команда по уходу рекомендовала французский метод, получите направление к физиотерапевту, который специализируется на этом методе.
Другие вопросы к врачу, если у вашего ребенка косолапость:
- Когда мой ребенок должен начать лечение?
- Какой лучший метод лечения косолапости у моего ребенка?
- Как долго будет продолжаться лечение?
- Будет ли мой ребенок нормально ходить?
- Как я могу предотвратить возвращение косолапости?
Справка из клиники Кливленда
Косолапость, также называемая эквиноварусной косолапостью, является распространенным врожденным дефектом.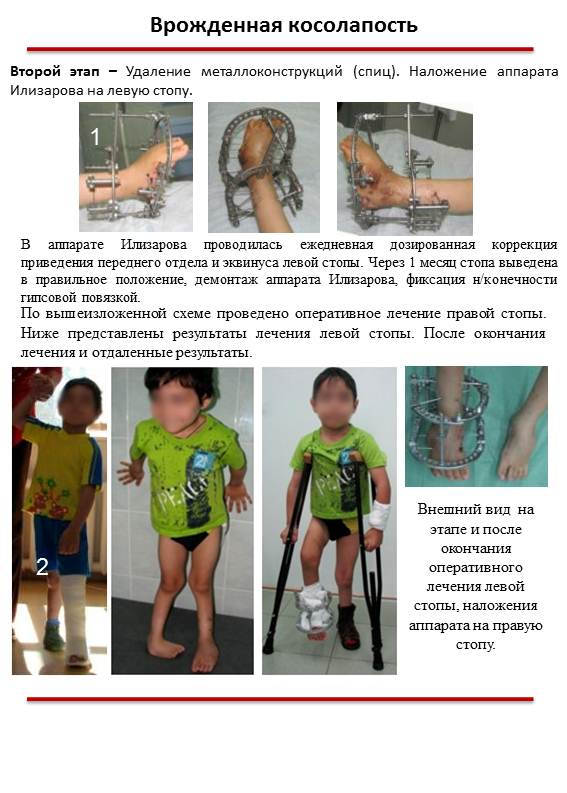 Нога или стопы ребенка поворачиваются внутрь. Косолапость не исчезнет сама по себе. Но лечение очень успешное. Терапия часто начинается в течение первых нескольких недель жизни. Нехирургические методы, такие как метод Понсети, могут вернуть стопу в правильное положение. Возможно, вашему ребенку в течение нескольких лет придется носить ортез для ног. Важно четко следовать схеме лечения. Это увеличивает шансы на успех. При правильном лечении многие дети с косолапостью могут безболезненно ходить, бегать и даже заниматься спортом. Поговорите со своим лечащим врачом о наилучшем методе лечения косолапости вашего ребенка.
Нога или стопы ребенка поворачиваются внутрь. Косолапость не исчезнет сама по себе. Но лечение очень успешное. Терапия часто начинается в течение первых нескольких недель жизни. Нехирургические методы, такие как метод Понсети, могут вернуть стопу в правильное положение. Возможно, вашему ребенку в течение нескольких лет придется носить ортез для ног. Важно четко следовать схеме лечения. Это увеличивает шансы на успех. При правильном лечении многие дети с косолапостью могут безболезненно ходить, бегать и даже заниматься спортом. Поговорите со своим лечащим врачом о наилучшем методе лечения косолапости вашего ребенка.
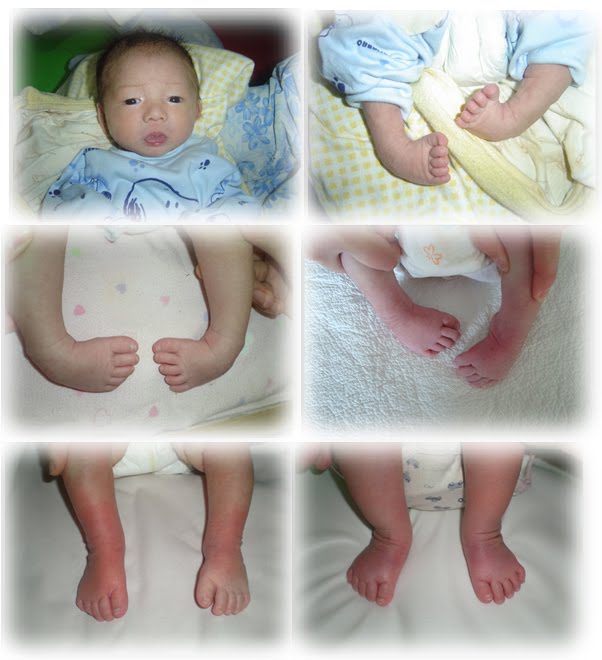
Глава 4.9. Эквиноварусная косолапость Эквиноварусная косолапость (Q66.0)
- Глава 4. Диагностика и кодирование врожденных аномалий
- Дефекты нервной трубки
- 4.2a Анэнцефалия
- 4.2b Краниорахишизис (Q00.1)
- 4.2c Иниэнцефалия (Q00.2)
- 4.2d Энцефалоцеле (Q01.0–Q01.83, Q01.9))
- 4.2e Spina Bifida (Q05.
 0–Q05.9)
0–Q05.9) - 4.3 Врожденные аномалии нервной системы: микроцефалия
- 4.4 Врожденные пороки развития уха
- 4.5b Общий ствол (Q20.0)
- 4.5c Транспозиция магистральных артерий (Q20.3)
- 4.5d Тетрада Фалло
- 4.5e Атрезия легочного клапана (Q22.0)
- 4.5f Атрезия трехстворчатого клапана (Q22.0) Q22.4)
- 4,5g Синдром гипоплазии левых отделов сердца (Q23.4)
- 4,5h Перерыв дуги аорты (q25.21, предпочтительнее; также Q25.2, Q25.4)
- 4.6 Орофациальные расщелины
- 4.7 Врожденные пороки развития пищеварительной системы
- 4.8 Врожденные аномалии половых органов Гипоспадия (Q54.0–Q54.9)
- 4.9a Врожденные аномалии и деформации опорно-двигательного аппарата: эквиноварусная косолапость (Q66.0)
- 4.9b Врожденные аномалии развития и деформации опорно-двигательного аппарата : Дефекты редукции конечностей/дефекты конечностей
- 4.9c Укорочение конечностей Амелия (Q71.0, Q72.
 0, Q73.0)
0, Q73.0) - 4.9d Укорочение конечностей: поперечный терминальный (Q71.2, Q71.3, Q71.30, Q72.2, Q72.3, Q72.30)
- 4.9e Недостаточность конечности: поперечная интеркалярная (Q71.1, Q72.1, Q72.4)
- 4.9f Недостаточность конечности: продольная преаксиальная (голень, лучевая кость, первый луч) (Q71.31, Q71. 4, Q72.31, Q72.5)
- 4.9g Дефицит конечностей: продольный постаксиальный (малоберцовая кость, локтевая кость, пятый луч) (Q71.30, Q71.5, Q72.30, Q72.6)
- 4.9h Дефицит конечностей : Дефицит продольной аксиальной конечности – разделение кисти и стопы (Q71.6, Q72.7)
- 4.10 Дефекты брюшной стенки
- 4.11 Хромосомные аномалии
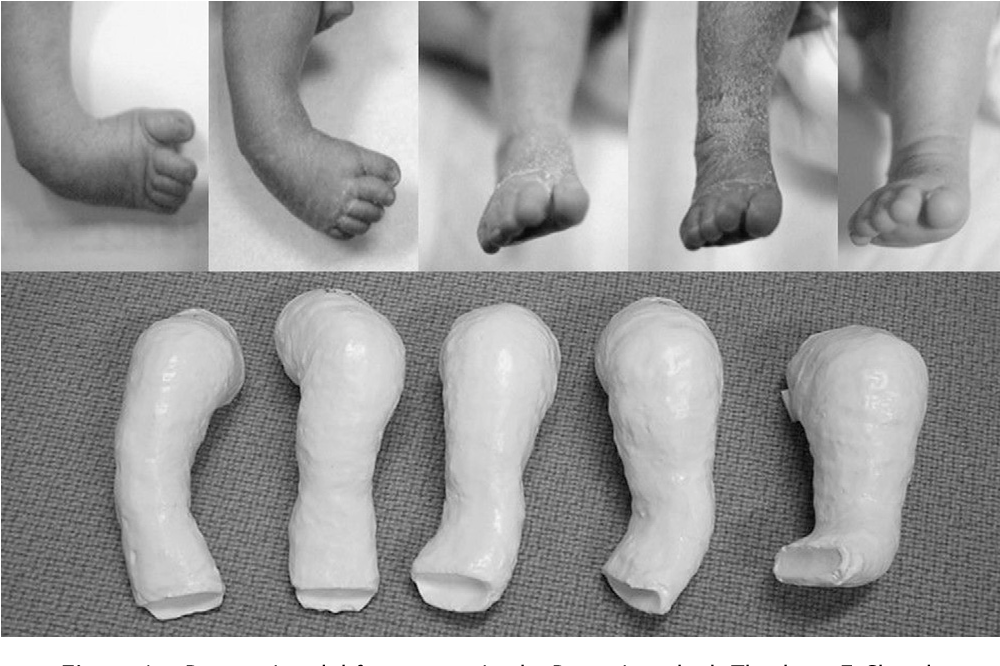
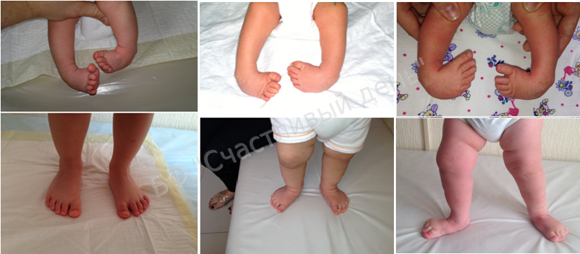
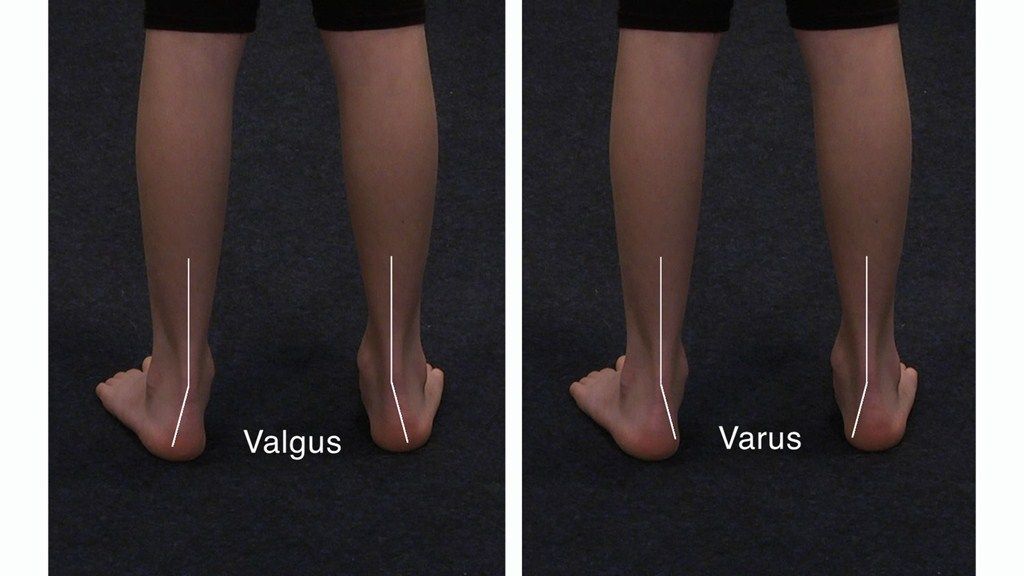
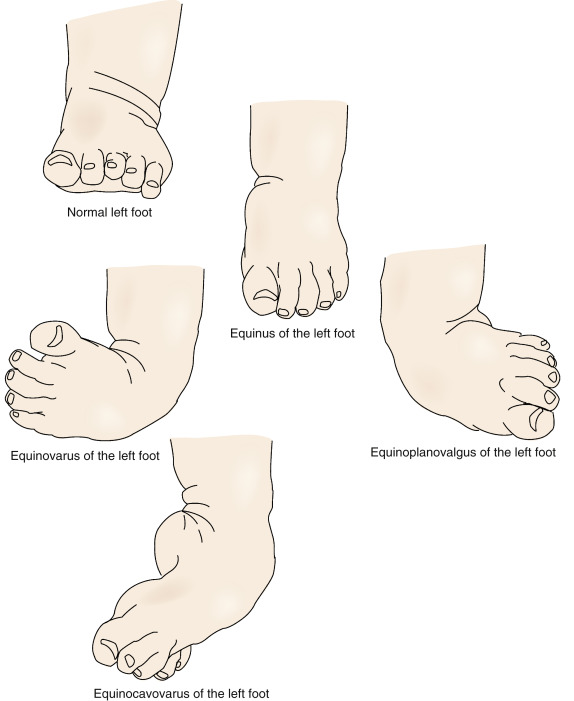
Эквиноварусная косолапость (TEV) представляет собой специфический и распространенный тип того, что иногда называют «косолапостью», термин, который охватывает ряд аномалий голеностопного сустава или стопы, присутствующих при рождении (см. рис. 4.33). TEV можно определить как фиксацию стопы (переднего и заднего отделов стопы) в подошвенном сгибании (эквинус), отклонении к средней линии (варус) и ротации вверх, так что стопа опирается на внешнюю сторону (супинат). Другими словами, стопа направлена вниз и внутрь и повернута наружу в осевом направлении, как показано на рис. 4.34.
Другими словами, стопа направлена вниз и внутрь и повернута наружу в осевом направлении, как показано на рис. 4.34.
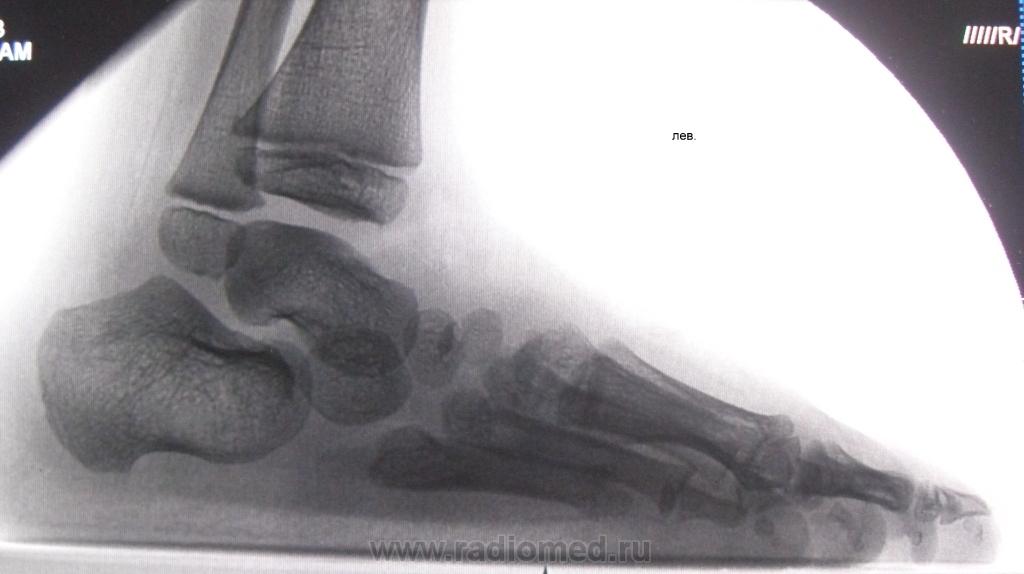
Рис. 4.33. Косолапость эквиноварусная
Источник фотографии и рентгеновского снимка: д-р Идалина Монтес и д-р Рафаэль Лонго (Пуэрто-Рико).
Рис. 4.34. Положения стоп
TEV имеет широкий спектр тяжести. В более легких случаях он является «позиционным», что означает, что его можно аккуратно придать нормальному положению и, как правило, не требует ортопедических или хирургических вмешательств, и он исключен из эпиднадзора за врожденными дефектами. В более тяжелых случаях он может быть «жестким» или «фиксированным», поскольку им нельзя манипулировать в нормальное положение, и он требует ортопедического или хирургического лечения и считается серьезным врожденным дефектом.
Наиболее частой врожденной деформацией стоп является ТЭВ; однако существуют и другие формы косолапости, в частности пяточно-вальгусная косолапость (при которой голеностопный сустав сгибается в тыльную сторону, а передний отдел стопы отклоняется наружу) и пяточно-пяточная косолапость (при которой голеностопный сустав сгибается в тыльную сторону, а передний отдел стопы отклоняется внутрь).
Соответствующие коды по МКБ-10
Q66.0 Эквиноварусная косолапость
Q66.8 Другие врожденные деформации стоп, косолапость БДУ (не указано иное)
Q66.1 Пяточно-варусная косолапость
Q66.4 Пяточно-вальгусная косолапость
Примечание:
Q66 Врожденные деформации стоп: избегайте использования этого общего кода, если доступна более конкретная информация.
Q66.8 Другие врожденные деформации стопы; Косолапость БДУ (не указано иное): по возможности сведите к минимуму использование этого кода; опишите аномалию, чтобы можно было использовать более конкретный код (например, Q66.0).
Диагностика
Пренатальная. Косолапость можно выявить или заподозрить на пренатальном УЗИ. Однако его не следует включать в данные эпиднадзора за врожденными пороками без постнатального подтверждения. Основная полезность пренатальной диагностики косолапости заключается в том, что она является показанием для дополнительных оценок генетических состояний и структурных аномалий, которые обычно связаны с TEV.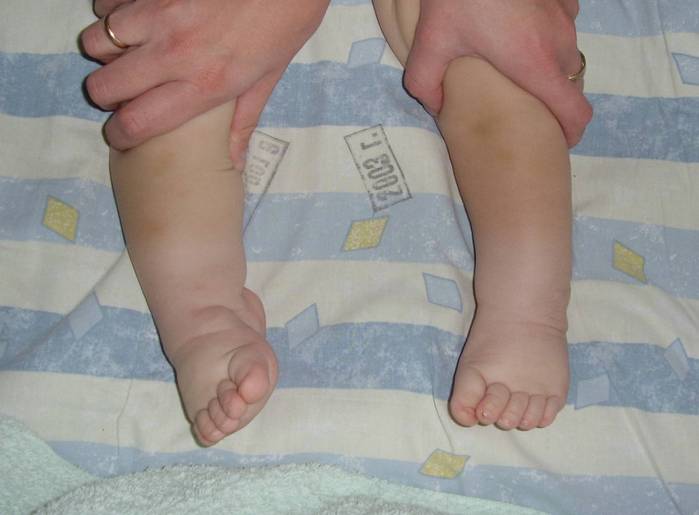
Послеродовой. Косолапость легко диагностируется при осмотре новорожденного. Случаи следует отслеживать и оценивать последовательно, чтобы оценить степень тяжести и необходимость лечения, отличного от манипуляций. Иногда другие врожденные дефекты стопы или ноги могут имитировать косолапость. Например, дефицит большеберцовой кости в ноге может выглядеть как косолапость. Визуализирующие исследования (как правило, рентгенограммы) могут предоставить дополнительную информацию, помогающую в диагностике.
Клинические и эпидемиологические примечания
TEV является двусторонним примерно в 60% случаев, а когда односторонний, TEV несколько чаще встречается справа. Особенно при тяжелых формах (фиксированный или ригидный ТЭВ) икроножные мышцы на стороне поражения гипотрофированы (уменьшены).
Примерно в половине всех случаев TEV возникает отдельно или в сочетании с другими связанными аномалиями опорно-двигательного аппарата, такими как кривошея, дисплазия тазобедренного сустава и аномалии множественных суставов (например, артрогрипоз).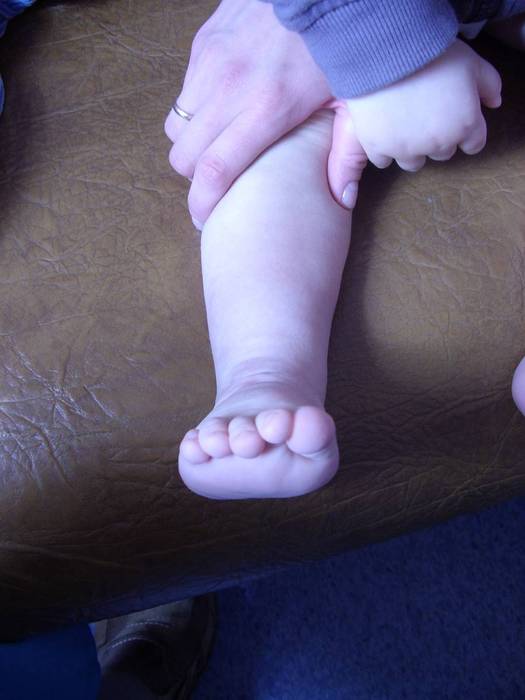 TEV может возникать при других врожденных дефектах, особенно тех, которые поражают головной мозг и позвоночник (например, расщепление позвоночника), посредством механизма, который, как считается, включает дефицит иннервации сегментов конечностей поперек суставов, что, в свою очередь, приводит к уменьшению движений внутриутробно. Множественные деформации, включающие TEV, могут возникать при генетических состояниях, влияющих на формирование костей и суставов (например, кампомелическая дисплазия, синдром Ларсена). Чаще TEV может возникать при хромосомных аномалиях, таких как триплоидия, делеция 4p- и трисомии (хотя при трисомии 18 тип косолапости пяточно-вальгусный, с тыльным, а не подошвенным сгибанием стопы).
TEV может возникать при других врожденных дефектах, особенно тех, которые поражают головной мозг и позвоночник (например, расщепление позвоночника), посредством механизма, который, как считается, включает дефицит иннервации сегментов конечностей поперек суставов, что, в свою очередь, приводит к уменьшению движений внутриутробно. Множественные деформации, включающие TEV, могут возникать при генетических состояниях, влияющих на формирование костей и суставов (например, кампомелическая дисплазия, синдром Ларсена). Чаще TEV может возникать при хромосомных аномалиях, таких как триплоидия, делеция 4p- и трисомии (хотя при трисомии 18 тип косолапости пяточно-вальгусный, с тыльным, а не подошвенным сгибанием стопы).
Негенетические факторы риска, которые, как сообщается, связаны с повышенным риском TEV, включают курение матери и, возможно, очень ранний амниоцентез. Кроме того, риск TEV, по-видимому, является многофакторным, поскольку риск рецидива у родственников первой степени родства составляет 3–6%, а конкордантность у монозиготных близнецов намного выше, чем у дизиготных близнецов (30% против примерно 3%).
Зарегистрированная распространенность при рождении составляет 10–15 на 10 000 рождений с умеренной вариабельностью. Такая изменчивость, вероятно, частично связана с методологией (определение, критерии включения), хотя этническая принадлежность, по-видимому, играет роль, с более высокими зарегистрированными показателями в определенных группах (например, маори, полинезийцы, австралийские аборигены).
Включения
Врожденная эквиноварусная косолапость (включая врожденную, идиопатическую и нейрогенную)
Косолапость, не уточненная иначе, косолапость, не уточненная иначе (свернуть эту группу) Пяточно-вальгусная косолапость
Q66.8 Другие врожденные деформации стоп, косолапость БДУ (не указано иное)
Исключения
Косолапость, позиционная (исключена; позиционная косолапость не считается серьезным дефектом)
Другие проявления врожденных деформаций стопы (например, варусная или вальгусная плюсневая кость, качающаяся стопа, плоская стопа, вогнутая стопа и т. д.)
д.)
- Латеральность – правая, левая или двусторонняя.
- Подвижность foo t – жесткая (сжатая) по сравнению с гибкой (гибкая такая же, как позиционная, и исключается в большинстве 9системы 0443).
- Типичные признаки эквиноварусной косолапости (подошвенное сгибание, варусная деформация, ротация вверх).
- Сопутствующие находки , если имеются (медиальные складки, гипотрофия голени).
- Опишите другие деформации, если они имеются (например, колени, пальцы, локти), особенно если они ригидны (например, при артрогрипозе).
- Опишите процедуры для оценки дальнейших дополнительных пороков развития; если присутствует один или несколько, опишите их.
- Сделать и сообщить о фотографиях аномалии и всего младенца; полезно для обзора, но недостаточно в качестве подтверждения.

- Консультации по специальности: Отчет о проделанной работе (включая генетику и ортопедию) и результаты.
- Обратите внимание на следующее:
- Гибкая/позиционная эквиноварусная косолапость или другие проявления деформации стопы исключены из наблюдения из-за изменчивости, частоты и незначительного воздействия на здоровье.
- Косолапость, связанная с нервно-мышечными нарушениями и синдромами, включена в отслеживание эпиднадзора; обратите внимание, что программы должны кодировать ассоциированную косолапость, но должны учитывать, включены ли эти случаи в оценки распространенности косолапости.
- Другие проявления деформации стопы.
Предлагаемые показатели качества данных
| Категория | Предлагаемые методы и показатели качества |
| Описание и документация | Обзор образца клинического описания для документирования ключевых дескрипторов:
|
| Кодировка |
|
| Клинический классификация |
|
| Распространенность |
|


Abstract
biorxivBIORXIVbioRxivbioRxivCold Spring Harbor Laboratory10.1101/402388biorxiv;402388v2biorxiv;4023884023884023884023881.2Regular ArticleNew ResultsGeneticsHomozygous TRPV4 mutation causes congenital distal spinal muscular atrophy and arthrogryposis*Corresponding Authors Vandana A Gupta, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital , Гарвардская медицинская школа, Бостон, Массачусетс, 02115, электронная почта: vgupta@research. bwh.harvard.edu; Рашель Годе, кафедра молекулярной и клеточной биологии, Гарвардский университет, Кембридж, Массачусетс, 02138, электронная почта: [email protected]#
bwh.harvard.edu; Рашель Годе, кафедра молекулярной и клеточной биологии, Гарвардский университет, Кембридж, Массачусетс, 02138, электронная почта: [email protected]#
Соавторы
Раскрытие финансовой информации
Г-н Велилья сообщает об отсутствии раскрытия информации.
Г-н Маркетти не сообщает о раскрытии информации.
Доктор Тот-Петроци не сообщает о раскрытии информации.
Мисс Гросгогит не сообщает о раскрытии информации.
Мисс Беннет не сообщает о раскрытии информации.
Мисс Кармайкл не сообщает о раскрытии информации.
Г-жа Эстрелла не сообщает о раскрытии информации.
Д-р Даррас получил гонорары как автор статей о нервно-мышечных заболеваниях для UpToDate, Inc. UpToDate не производит продукты или услуги, связанные со здравоохранением. Доктор Даррас работал консультантом (членом специального научного консультативного совета) в компаниях Sarepta Inc. , AveXis, Inc., BMS, Inc., Cytokinetics, Inc., Biogen, Inc., Marathon, Inc., Santhera, Inc., и Рош, Инк.; получает исследовательскую поддержку от PTC, Inc., Summit, Inc., Fibrogen, Inc., Santhera, Inc., Cyokinetics, Inc., Biogen, Inc., AveXis, Inc., Roche, Inc., Национальных институтов здравоохранения ( НИНДС, 5У10НС077269), Фонд SMA, Фонд Working on Walking и Семейный фонд Slaney для SMA. У доктора Дарраса нет конфликта интересов в отношении этой рукописи.
, AveXis, Inc., BMS, Inc., Cytokinetics, Inc., Biogen, Inc., Marathon, Inc., Santhera, Inc., и Рош, Инк.; получает исследовательскую поддержку от PTC, Inc., Summit, Inc., Fibrogen, Inc., Santhera, Inc., Cyokinetics, Inc., Biogen, Inc., AveXis, Inc., Roche, Inc., Национальных институтов здравоохранения ( НИНДС, 5У10НС077269), Фонд SMA, Фонд Working on Walking и Семейный фонд Slaney для SMA. У доктора Дарраса нет конфликта интересов в отношении этой рукописи.
Д-р Франк является соавтором патентов США, связанных с ABCB5, переданных Brigham and Women’s Hospital, Бостон, Массачусетс, и лицензированных Ticeba GmbH (Гейдельберг, Германия) и Rheacell GmbH & Co. KG (Гейдельберг, Германия) . Она также является советником Veritas Genetics, Inc. (Данверс, Массачусетс). Ее супруг, доктор Маркус Франк, является научным консультантом Ticeba GmbH и Rheacell GmbH & Co. KG.
Доктор Криер не сообщает о раскрытии информации.
Доктор Годе не сообщает о раскрытии информации.
Д-р Гупта получил поддержку K01 AR062601 (VAG) и стипендии Элеоноры и Майлза Шор для ученых в области медицины, а также премии Brigham and Women’s Hospital Career Development Award (VAG).
VelillaJoseBS1#MarchettiMichael MarioBS2#Toth-PetroczyAgnesPhD2GrosgogeatClaireBS2BennettAlexis HBS2CarmichaelNikkolaMS, CGC2EstrellaEliciaMS, CGC3DarrasBasil T.MD4Brigham Genomic Medicine2FrankNatasha YMD2KrierJoelMD2GaudetRachellePhD1*GuptaVandana A.PhD2*1Department of Molecular and Cellular Biology, Harvard University, Cambridge, MA 021382Division of Genetics, Brigham and Women’s Hospital, Harvard Medical Школа, Бостон, Массачусетс 021153 Отделение генетики, Бостонская детская больница, Бостон, Массачусетс 021154 Отделение неврологии, Бостонская детская больница, Гарвардская медицинская школа, Бостон, Массачусетс 02115201829820181112201829820181112201840238828820181112201811122018© 2018, Опубликовано Cold Spring Harbor Laboratory2018
Правообладателем этого препринта является автор. Все права защищены. Материал не может распространяться, повторно использоваться или адаптироваться без разрешения автора.
Все права защищены. Материал не может распространяться, повторно использоваться или адаптироваться без разрешения автора.
AbstractObjective
Целью данного исследования является выявление генетической причины заболевания при врожденной форме врожденной спинальной мышечной атрофии и артрогрипоза (ВСМАА).
Методы
У 2-летнего мальчика был диагностирован врожденный множественный артрогрипоз, тяжелые скелетные аномалии, кривошея, паралич голосовых связок и снижение подвижности нижних конечностей. Полное экзомное секвенирование было проведено у пробанда и членов его семьи. Для подтверждения патогенности идентифицированного варианта было выполнено in silico моделирование структуры белка и экспрессии гетерологичного белка, а также анализы цитотоксичности.
Результаты
Полное секвенирование экзома выявило гомозиготную мутацию в гене TRPV4 (c.281C>T; p.S94L). Выявление рецессивной мутации в TRPV4 расширяет спектр мутаций при рецессивных формах заболевания, связанного с TRPV4. p.S94L и другие ранее идентифицированные варианты TRPV4 в различных белковых доменах сравнивали при структурном моделировании и функциональных исследованиях. Структурное моделирование in silico предполагает, что мутация p.S94L находится в неупорядоченной N-концевой области, проксимальной к важным регуляторным сайтам связывания фосфоинозитидов и PACSIN3, что может привести к изменениям в доставке и/или чувствительности каналов. Функциональные исследования вестерн-блоттингом и иммуногистохимическим анализом показывают, что p.S94L снижает стабильность белка TRPV4 из-за повышенной цитотоксичности и, следовательно, включает механизм усиления функции.
p.S94L и другие ранее идентифицированные варианты TRPV4 в различных белковых доменах сравнивали при структурном моделировании и функциональных исследованиях. Структурное моделирование in silico предполагает, что мутация p.S94L находится в неупорядоченной N-концевой области, проксимальной к важным регуляторным сайтам связывания фосфоинозитидов и PACSIN3, что может привести к изменениям в доставке и/или чувствительности каналов. Функциональные исследования вестерн-блоттингом и иммуногистохимическим анализом показывают, что p.S94L снижает стабильность белка TRPV4 из-за повышенной цитотоксичности и, следовательно, включает механизм усиления функции.
Заключение
Это исследование идентифицирует новую гомозиготную мутацию в TRPV4 как причину рецессивной формы врожденной спинальной мышечной атрофии и артрогрипоза.
особое-свойствосодержит-встроенный-дополнительный-материалимеет-более раннюю-версиюдаГлоссарийdHMN
дистальная наследственная моторная невропатия
HMSN
наследственная моторная и сенсорная невропатия
HSAN
hereditary sensory and autonomic neuropathy
SPSMA
scapuloperoneal spinal muscular atrophy
CSMAA
congenital spinal muscular atrophy and arthrogryposis
CMT
Charcot-Marie-Tooth diseases
PBD
phosphoinositide-binding domain
PRR
proline-rich region
PIP 2
фосфатидилинозитол 4,5-бифосфат
FDAB
Семейная пальцевая артропатия-брахидактилия
Сводка обновлений:
Рисунки 3 и 4 пересмотрены
Наследственные невропатии представляют собой клинически и генетически гетерогенную группу заболеваний с предполагаемой распространенностью 1:2500. Клинические проявления наследственных невропатий включают медленно прогрессирующую дистальную слабость и атрофию мышц с потерей чувствительности или без нее. В зависимости от клинического фенотипа наследственные невропатии подразделяются на три широкие категории. Болезнь Шарко-Мари-Тута (ШМТ) или наследственная моторная и сенсорная невропатия (HMSN) обычно демонстрируют поражение как моторной, так и сенсорной систем, наследственная сенсорная и вегетативная невропатия (HSAN) включает сенсорный дефицит и/или вегетативную дисфункцию, а также дистальную наследственную моторную невропатию. (dHMN) преимущественно связан с моторным дефицитом. Эти группы можно далее разделить на множество подтипов в зависимости от электрофизиологических критериев, патологических дефектов, типа наследования и молекулярно-генетических дефектов. Эти заболевания генетически очень гетерогенны с мутациями по крайней мере в 80 различных генах, связанных с этими подтипами 1, 2 . Несмотря на этот прогресс, 30-70% людей, страдающих невропатиями, не имеют генетического диагноза из-за клинической и генетической гетерогенности.
Клинические проявления наследственных невропатий включают медленно прогрессирующую дистальную слабость и атрофию мышц с потерей чувствительности или без нее. В зависимости от клинического фенотипа наследственные невропатии подразделяются на три широкие категории. Болезнь Шарко-Мари-Тута (ШМТ) или наследственная моторная и сенсорная невропатия (HMSN) обычно демонстрируют поражение как моторной, так и сенсорной систем, наследственная сенсорная и вегетативная невропатия (HSAN) включает сенсорный дефицит и/или вегетативную дисфункцию, а также дистальную наследственную моторную невропатию. (dHMN) преимущественно связан с моторным дефицитом. Эти группы можно далее разделить на множество подтипов в зависимости от электрофизиологических критериев, патологических дефектов, типа наследования и молекулярно-генетических дефектов. Эти заболевания генетически очень гетерогенны с мутациями по крайней мере в 80 различных генах, связанных с этими подтипами 1, 2 . Несмотря на этот прогресс, 30-70% людей, страдающих невропатиями, не имеют генетического диагноза из-за клинической и генетической гетерогенности.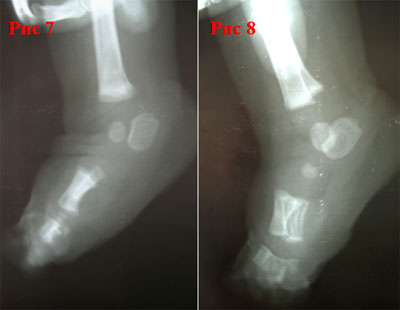 Некоторые симптомы болезни можно контролировать с помощью непатентованных препаратов. Тем не менее, идентификация основного генетического поражения необходима для точного прогноза заболевания, лечения и планирования семьи и может в конечном итоге привести к индивидуальному лечению.
Некоторые симптомы болезни можно контролировать с помощью непатентованных препаратов. Тем не менее, идентификация основного генетического поражения необходима для точного прогноза заболевания, лечения и планирования семьи и может в конечном итоге привести к индивидуальному лечению.
Мутации в гене катионного канала TRPV4 (транзиентный рецепторный потенциал ваниллоида 4) являются редкой причиной доминантно-наследственных аксональных невропатий и скелетных дисплазий 3, 4 . Мутации TRPV4 лежат в основе широкого спектра клинических проявлений и связаны с dHMN, лопаточно-перонеальной спинальной мышечной атрофией (SPSMA), врожденной спинальной мышечной атрофией и артрогрипозом (CSMAA), аутосомно-доминантным аксональным ШМТ типа 2C и врожденной дистальной спинальной мышечной атрофией (dSMA). Нейропатии, связанные с TRPV4, часто связаны с параличом голосовых связок и иногда с нейросенсорной тугоухостью. Мутации TRPV4 также могут приводить к аутосомно-доминантным скелетным дисплазиям. Как правило, различные мутации в гене TRPV4 связаны либо с невропатиями, либо со скелетной дисплазией; однако у некоторых пациентов проявляются оба клинических фенотипа. На сегодняшний день у пациентов с невропатиями идентифицировано более 20 различных мутаций TRPV4. Однако функциональное подтверждение патогенности многих из этих вариантов все еще отсутствует, что препятствует окончательному генетическому диагнозу у этих пациентов. Чтобы улучшить генетическую диагностику пациентов с невропатиями, мы используем полноэкзомное секвенирование (WES) в сочетании с функциональными проверочными исследованиями для установления патогенности вариантов, идентифицированных с помощью WES. В этой работе была выявлена гомозиготная мутация TRPV4 у пациента с дистальной наследственной моторной нейропатией. Последующие функциональные исследования выявляют различные механизмы заболевания для различных вариантов TRPV4 и дают новые идеи, которые могут помочь в разработке будущих терапевтических стратегий для этих пациентов.
Как правило, различные мутации в гене TRPV4 связаны либо с невропатиями, либо со скелетной дисплазией; однако у некоторых пациентов проявляются оба клинических фенотипа. На сегодняшний день у пациентов с невропатиями идентифицировано более 20 различных мутаций TRPV4. Однако функциональное подтверждение патогенности многих из этих вариантов все еще отсутствует, что препятствует окончательному генетическому диагнозу у этих пациентов. Чтобы улучшить генетическую диагностику пациентов с невропатиями, мы используем полноэкзомное секвенирование (WES) в сочетании с функциональными проверочными исследованиями для установления патогенности вариантов, идентифицированных с помощью WES. В этой работе была выявлена гомозиготная мутация TRPV4 у пациента с дистальной наследственной моторной нейропатией. Последующие функциональные исследования выявляют различные механизмы заболевания для различных вариантов TRPV4 и дают новые идеи, которые могут помочь в разработке будущих терапевтических стратегий для этих пациентов.
Материалы и методы Утверждения стандартного протокола, регистрации и согласия пациентов
Пробанд, оба родителя и незатронутые братья и сестры были включены в исследование, и от участников было получено информированное согласие в соответствии с одобренным Институциональным наблюдательным советом исследованием в Бостонской детской больнице и Бригаме и Женской больнице .
Секвенирование полного экзома
Экстракцию ДНК из образцов крови выполняли в исследовательском центре Biobank Core (Boston Children’s Hospital) с использованием мини-набора QIAmp DNA (Qiagen). Полное секвенирование экзома было выполнено Йельским центром генома. Образцы ДНК пробанда и родителей были отправлены на полноэкзомное секвенирование. Образцы были подготовлены в виде библиотеки для секвенирования Illumina и обогащены экзомными последовательностями с использованием набора Agilent V5 Sureselect. Захваченные библиотеки секвенировали с использованием секвенаторов Illumina HiSeq 2000 в Lab Corp. FASTQ, созданные в результате секвенирования экзома, фильтровали и выравнивали, а варианты фильтровали и аннотировали, как описано ранее 5 . Короче говоря, сначала мы применяем агностическую фильтрацию и используем фильтрацию режима наследования на основе родословной для de novo и рецессивных вариантов, а также фильтрацию только для редких вариантов на основе больших баз данных населения (gnomAD, gnomad.broadinstitute.org). На втором этапе мы применяем несколько основанных на знаниях фильтров к генам (например, известные функциональные ассоциации и ассоциации заболеваний, данные об уровне экспрессии, данные модели организма) и к вариантам (например, эволюционная консервация, структурные ограничения и т. д.). Наконец, варианты были расставлены по приоритетам во время краудсорсинговой конференции междисциплинарной аудитории. 5 Для проверки варианта TRPV4 продукты ПЦР для пробанда, здоровых братьев и сестер и родителей анализировали с помощью стандартного секвенирования по Сэнгеру (Dana-Farber/Harvard Cancer Center DNA Sequencing Facility).
FASTQ, созданные в результате секвенирования экзома, фильтровали и выравнивали, а варианты фильтровали и аннотировали, как описано ранее 5 . Короче говоря, сначала мы применяем агностическую фильтрацию и используем фильтрацию режима наследования на основе родословной для de novo и рецессивных вариантов, а также фильтрацию только для редких вариантов на основе больших баз данных населения (gnomAD, gnomad.broadinstitute.org). На втором этапе мы применяем несколько основанных на знаниях фильтров к генам (например, известные функциональные ассоциации и ассоциации заболеваний, данные об уровне экспрессии, данные модели организма) и к вариантам (например, эволюционная консервация, структурные ограничения и т. д.). Наконец, варианты были расставлены по приоритетам во время краудсорсинговой конференции междисциплинарной аудитории. 5 Для проверки варианта TRPV4 продукты ПЦР для пробанда, здоровых братьев и сестер и родителей анализировали с помощью стандартного секвенирования по Сэнгеру (Dana-Farber/Harvard Cancer Center DNA Sequencing Facility).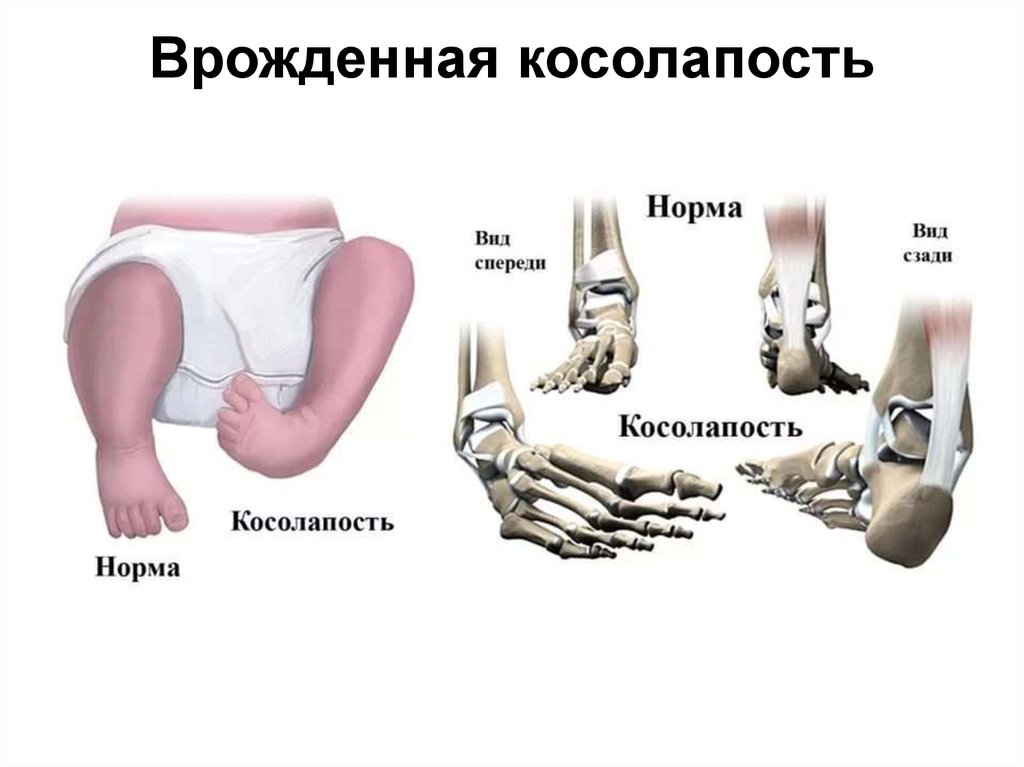
Моделирование in silico мутаций TRPV4
Мутации TRPV4 были картированы на криоЭМ-структуре Xenopus tropicalis TRPV4 (идентификатор PDB: 6bjj) 6 после выравнивания последовательностей XtTRPV4 и TRPV4 человека с помощью Clustal Omega. Рисунок 2 был создан с использованием PyMOL (Schrodinger).
biorxiv;402388v2/FIG2F1fig2Рис. 2.
Структурная модель, основанная на криоЭМ-структуре Xenopus tropicalis TRPV4 (идентификатор PDB: 6bjj). Структура, соответствующая остаткам 148-788 (человеческая нумерация), не включает неупорядоченные N- и С-концевые участки. N-концевая область схематически изображена пунктирной линией для каждой субъединицы с указанием фосфоинозитид-связывающего домена (PBD; остатки 121-125 у человека) и области, богатой пролином (PRR, остатки 135-144). Положения остатков для мутаций, вызывающих невропатию (D62N, P97R, R186Q, R232C/S, R237G/L, R269C/H, R315W, R316C/H, T701I) и мутации болезни со смешанными фенотипами (G78W, A217S, E278K, S542Y, V620Y, T740I) отмечены зеленым и желтым цветом соответственно. Положение p.S94L указано фиолетовым цветом.
Положение p.S94L указано фиолетовым цветом.
Клонирование мутантных конструкций
Для создания мутантных плазмид TRPV4 варианты p.S94L, p.R315W и & p.T701I были включены в кДНК TRPV4 в плазмиде pcDNA3.1-TRPV4-FLAG с использованием набора для направленного мутагенеза Q5 (Новая Англия). биолаборатории). Последовательности праймеров для мутагенеза были следующими: S94L вперед: 5’-TAT GAG TCC TTG GTG GTG CCT-3’, S94L реверс: 5′-TAG GGT GGA CTC CAG CAG-3′, R315W вперед: 5′-GGC GGA CAT GTG GCG CCA GGA-3′, R315W реверс: 5′-TTC TTG TGG GGG TTC TCC GTC AGGT-3′ , T701I вперед: 5′-CTG CTG GTG ATC TAC ATC ATC-3′, T701I назад: 5′-GAT GAT GAA GAC CAC GGG-3′. Полные кодирующие последовательности были подтверждены с помощью секвенирования по Сэнгеру.
Анализ цитотоксичности
Клетки HEK293T человека культивировали в среде DMEM с добавлением 10% эмбриональной бычьей сыворотки. Клетки трансфицировали соответствующим TRPV4 или пустым вектором с использованием Lipofectamine 3000 (Thermo Fisher Scientific) и культивировали в присутствии или в отсутствие HC-067047 (5 мкМ). Анализ гибели клеток проводили через 24 часа после трансфекции с использованием набора для обнаружения цитотоксичности (Roche Diagnostics, Индианаполис, Индиана) в соответствии с инструкциями производителя. Цитотоксичность рассчитывали путем вычитания оптической плотности фонового контроля (среда с пустым вектором) из оптической плотности экспериментальных образцов (супернатант клеток, трансфицированных WT или мутантной плазмидой).
Анализ гибели клеток проводили через 24 часа после трансфекции с использованием набора для обнаружения цитотоксичности (Roche Diagnostics, Индианаполис, Индиана) в соответствии с инструкциями производителя. Цитотоксичность рассчитывали путем вычитания оптической плотности фонового контроля (среда с пустым вектором) из оптической плотности экспериментальных образцов (супернатант клеток, трансфицированных WT или мутантной плазмидой).
Вестерн-блоттинг
Клетки HEK293 трансфицировали соответствующим вектором с использованием полиэтиленимина (соотношение PEI к ДНК 3:1). Клетки инкубировали в течение 12 часов при 37°С, затем доводили до 33°С и собирали через 48 часов после трансфекции. Вестерн-блоты выполняли в соответствии со стандартными протоколами, используя 1:2000 анти-FLAG M2 (Sigma, F-3165) для обнаружения TRPV4, 1:10000 анти-GAPDH (Abcam, 8245) и 1:1000 конъюгированные щелочной фосфатазой антимышиные антитела. вторичное антитело (Sigma, A3562). Мембраны разрабатывали с использованием реагента 1-Step NBT/BCIP (ThermoFisher Scientific, 34042).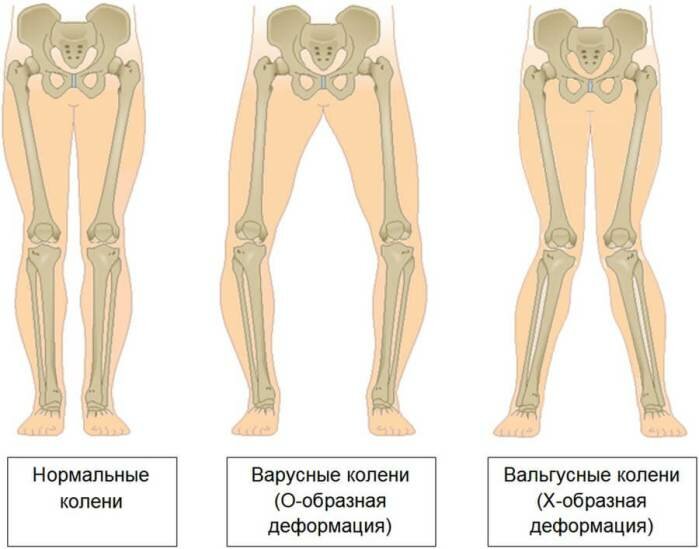 Денситометрический анализ проводили с помощью imageJ (https://imagej.nih.gov/ij/).
Денситометрический анализ проводили с помощью imageJ (https://imagej.nih.gov/ij/).
Иммунофлуоресцентный анализ
Для проведения иммунофлуоресценции клетки HEK293T высевали на 8-камерные предметные стекла и проводили трансфекцию липофектамином 3000 (Thermo Fisher Scientific). Через 24 часа после трансфекции клетки фиксировали в 4% параформальдегиде и проводили иммунофлуоресценцию, как описано ранее 16 . Антитело М2 к FLAG (Sigma, 1:250) и антитело к натрий-калиевой АТФазе (Abcam 76020, 1:100). Окрашивание ядер проводили с использованием DAPI (Biolegend, 422801).
РезультатыКлиническая картина
Пробанд-мужчина родился от неродственных здоровых родителей пуэрториканского происхождения после 37 недель беременности (рис. 1А). У него есть здоровая сестра. Пробанд родоразрешен путем кесарева сечения в связи с тазовым предлежанием и врожденным множественным артрогрипозом на УЗИ. При рождении у пробанда была обнаружена правая косолапость, врожденная левая вертикальная таранная кость и двусторонние сгибательные деформации коленей. Рентген показал вывих бедра с широким проксимальным отделом бедренной кости. УЗИ показало дисплазию вертлужных впадин с обеих сторон. Было сделано УЗИ позвоночника, которое показало, что спинной мозг находится на уровне L3. Двусторонняя гибкая ларингоскопия была выполнена через левую ноздрю, которая показала необструктивную гипертрофию аденоидов, нормальное основание языка, структурно нормальную гортань без признаков ларингомаляции. Голосовые связки были хорошо видны и казались неподвижными с обеих сторон в парамедианном положении. У пробанда также наблюдался инспираторный стридор. При осмотре правой и левой барабанных перепонок были обнаружены двусторонние выпоты в среднем ухе, нормальное состояние наружного уха и наружного слухового прохода. Аудиометрия с визуальным подкреплением показала умеренную кондуктивную тугоухость как минимум на одно ухо на частотах 500 и 2000 Гц. Тимпанограммы были плоскими с обеих сторон, что соответствовало клиническим данным.
Рентген показал вывих бедра с широким проксимальным отделом бедренной кости. УЗИ показало дисплазию вертлужных впадин с обеих сторон. Было сделано УЗИ позвоночника, которое показало, что спинной мозг находится на уровне L3. Двусторонняя гибкая ларингоскопия была выполнена через левую ноздрю, которая показала необструктивную гипертрофию аденоидов, нормальное основание языка, структурно нормальную гортань без признаков ларингомаляции. Голосовые связки были хорошо видны и казались неподвижными с обеих сторон в парамедианном положении. У пробанда также наблюдался инспираторный стридор. При осмотре правой и левой барабанных перепонок были обнаружены двусторонние выпоты в среднем ухе, нормальное состояние наружного уха и наружного слухового прохода. Аудиометрия с визуальным подкреплением показала умеренную кондуктивную тугоухость как минимум на одно ухо на частотах 500 и 2000 Гц. Тимпанограммы были плоскими с обеих сторон, что соответствовало клиническим данным.
biorxiv;402388v2/FIG1F2fig1Рис.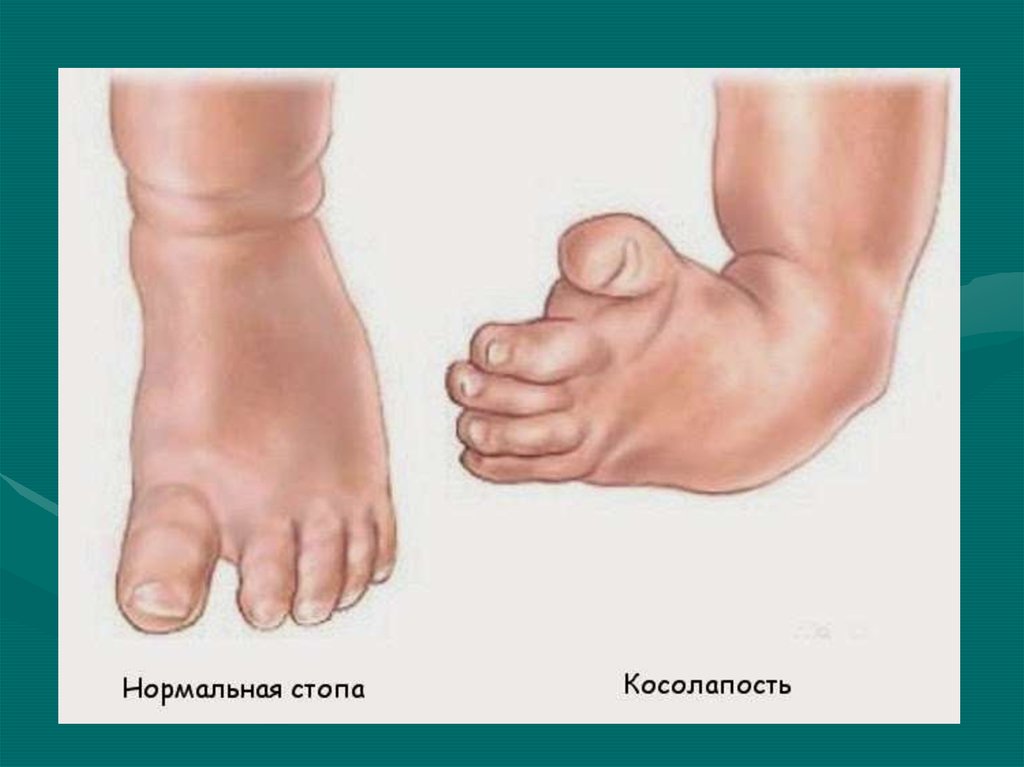 1.
1.
(A) Родословная семьи больного пациента; пробанд обозначен стрелкой (B). Ферограммы секвенирования по Сэнгеру показывают гомозиготность по c.281C>T в TRPV4 у пробанда и гетерозиготность у обоих родителей. (C) Выравнивание белковых последовательностей ортологов TRPV4, показывающее консервативность в области, включая p.S94L, у высших позвоночных. (D) Схематическая диаграмма белка TRPV4, демонстрирующая локализацию p.S94L в N-концевой межклеточной области. Номера 1-6 соответствуют шести анкириновым повторам.
В возрасте 2 лет пробанд мог самостоятельно сидеть и ходить на коленях. Однако он не мог стоять или ходить самостоятельно. За исключением движений нижних конечностей, пробанд имеет нормальную речь, мелкую и крупную моторику верхних конечностей, нормальное развитие слуха и зрения и демонстрирует статическое состояние.
Электромиография (ЭМГ) показала нормальную сенсорную реакцию справа и нормальную сенсорную реакцию левой локтевой кости. Также была отмечена нормальная для данного возраста двигательная реакция левой большеберцовой АГ и срединной APB. Концентрическое игольчатое исследование выбранных мышц показало потенциалы фибрилляции в правой латеральной широкой мышце с поздними и быстрыми потенциалами двигательных единиц в левой дельтовидной, правой передней большеберцовой и правой подвздошно-поясничной мышцах. Электрофизиологические данные свидетельствуют о генерализованной моторной аксонопатии с сосуществующими изменениями денервации и реиннервации.
Также была отмечена нормальная для данного возраста двигательная реакция левой большеберцовой АГ и срединной APB. Концентрическое игольчатое исследование выбранных мышц показало потенциалы фибрилляции в правой латеральной широкой мышце с поздними и быстрыми потенциалами двигательных единиц в левой дельтовидной, правой передней большеберцовой и правой подвздошно-поясничной мышцах. Электрофизиологические данные свидетельствуют о генерализованной моторной аксонопатии с сосуществующими изменениями денервации и реиннервации.
Полное секвенирование экзома (WES)
Полное секвенирование экзома проводили у пробанда и родителей. Присутствие единственного больного мужского пола в этой семье согласуется с аутосомно-рецессивным наследованием, Х-сцепленным наследованием или с доминантной мутацией de novo. Поэтому была проведена фильтрация на основе родословной и популяции для выявления потенциально опасных гомозиготных, Х-сцепленных, сложных гетерозиготных и de novo вариантов. В базе данных gnomAD (gnomad.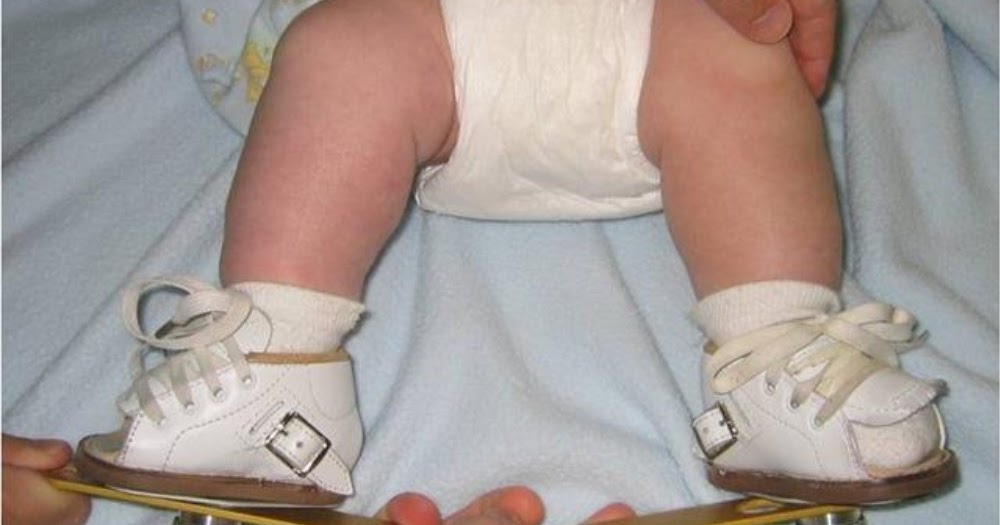 broadinstitute.org) в зависимости от доминантного или рецессивного типа наследования, что согласуется с распространенностью заболевания, мы рассматривали только редкие варианты кодирования и сплайсинга, которые имеют <0,1-1 % аллелей в общей здоровой популяции в общей здоровой популяции. В общей сложности 15 потенциальных генов содержали варианты (таблица e-1), которым был присвоен дополнительный приоритет с использованием нашего основанного на знаниях и краудсорсингового конвейера, недавно рассмотренного Haghighi et al. 5 . Главным кандидатом был гомозиготный миссенс-вариант в TRPV4 c.281C>T; (стр.S94L). Секвенирование по Сэнгеру подтвердило гомозиготность этого варианта у пробанда (BGM0049-1) и гетерозиготное наследование от обоих родителей (BGM0049-2, -3) (рис. 1Б). Было обнаружено, что здоровая сестра гомозиготна по нормальному варианту (не показано). Другие 14 вариантов/генов можно было бы исключить из-за низкой ограниченности популяции, известных ассоциаций неродственных заболеваний, неродственных функций белков и отсутствия экспрессии в соответствующих тканях (таблица e-1), что подтверждает потенциальный причинный эффект варианта TRPV4.
broadinstitute.org) в зависимости от доминантного или рецессивного типа наследования, что согласуется с распространенностью заболевания, мы рассматривали только редкие варианты кодирования и сплайсинга, которые имеют <0,1-1 % аллелей в общей здоровой популяции в общей здоровой популяции. В общей сложности 15 потенциальных генов содержали варианты (таблица e-1), которым был присвоен дополнительный приоритет с использованием нашего основанного на знаниях и краудсорсингового конвейера, недавно рассмотренного Haghighi et al. 5 . Главным кандидатом был гомозиготный миссенс-вариант в TRPV4 c.281C>T; (стр.S94L). Секвенирование по Сэнгеру подтвердило гомозиготность этого варианта у пробанда (BGM0049-1) и гетерозиготное наследование от обоих родителей (BGM0049-2, -3) (рис. 1Б). Было обнаружено, что здоровая сестра гомозиготна по нормальному варианту (не показано). Другие 14 вариантов/генов можно было бы исключить из-за низкой ограниченности популяции, известных ассоциаций неродственных заболеваний, неродственных функций белков и отсутствия экспрессии в соответствующих тканях (таблица e-1), что подтверждает потенциальный причинный эффект варианта TRPV4. Этот вариант миссенс высоко консервативен у большинства видов позвоночных и локализован на N-конце белка TRPV4 (Fig. 1C-D).
Этот вариант миссенс высоко консервативен у большинства видов позвоночных и локализован на N-конце белка TRPV4 (Fig. 1C-D).
Структурная модель
Недавняя криоэлектронная микроскопия (криоЭМ) структуры Xenopus tropicalis TRPV4 была использована для моделирования положения p.S94L и других ранее описанных мутаций TRPV4, вызывающих невропатию (рис. 2) 6 . В соответствии с предыдущей защитой от протеолиза 7 и исследованиями ядерного магнитного резонанса 8 N-концевая область — до остатка 147 (человеческая нумерация), таким образом, включая p.S94 — была разупорядочена и не моделировалась в криоЭМ-структуре. Однако стр.S94L расположен на N-конце по отношению к двум важным регуляторным областям: фосфоинозитид-связывающему домену (PBD; остатки 121-125 у человека), который взаимодействует с PIP 2 7 , и области, богатой пролином (PRR; остатки 135-144). у людей), который взаимодействует с доменом Sh4 PACSIN3 9 . PACSIN3 усиливает доставку TRPV4 к клеточной мембране. Связывание PIP 2 усиливает ответы TRPV4 на несколько стимулов, тогда как PACSIN3 снижает ответы на те же самые стимулы посредством антагонистического взаимодействия между двумя ключевыми регуляторами, PIP 2 и PACSIN3, что, вероятно, опосредовано опосредованными PACSIN3 структурными перестройками PRR 7, 10 . Следовательно, наше структурное моделирование предполагает, что мутация p.S94L потенциально может влиять на регуляцию и/или чувствительность TRPV4.
PACSIN3 усиливает доставку TRPV4 к клеточной мембране. Связывание PIP 2 усиливает ответы TRPV4 на несколько стимулов, тогда как PACSIN3 снижает ответы на те же самые стимулы посредством антагонистического взаимодействия между двумя ключевыми регуляторами, PIP 2 и PACSIN3, что, вероятно, опосредовано опосредованными PACSIN3 структурными перестройками PRR 7, 10 . Следовательно, наше структурное моделирование предполагает, что мутация p.S94L потенциально может влиять на регуляцию и/или чувствительность TRPV4.
Функциональное моделирование вариантов TRPV4
Функциональные исследования показали механизм приобретения функции для ранее известных доминантных мутаций в TRPV4 11 . Для подтверждения патогенности варианта p.S94L были проведены функциональные исследования путем анализа стабильности и локализации мутантного белка. Чтобы сравнить величину функционального дефицита, известный вызывающий невропатию p.R269Мутанты C и p.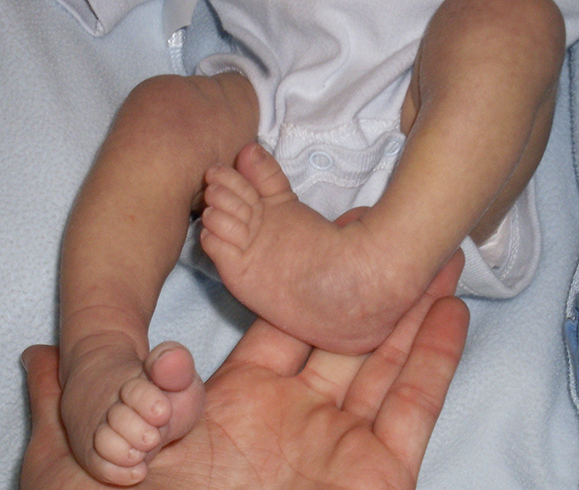 R315W TRPV4 анализировали параллельно. Кроме того, сообщалось, что мутация p.T701I является причиной невропатии. p.T701I локализован в трансмембранном домене спирали S6 (рис. 2), рядом с сайтом связывания ваниллоида в гомологичном ионном канале TRPV1. 12 В настоящее время нет функциональных исследований, устанавливающих патогенность этого варианта при заболеваниях человека или функции белка TRPV4. Поэтому мы исследовали влияние p.S94L, p.R269C, p.R314W и p.T701I на стабильность и субклеточную локализацию TRPV4. Чтобы исследовать влияние различных вариантов на стабильность белка, TRPV4-WT и мутантные конструкции были временно экспрессированы в HEK29.3T-клетки и их экспрессию анализировали с помощью вестерн-блоттинга (фиг. 3). Анализ белка с помощью вестерн-блоттинга показал снижение уровня белка p.S94L, а также снижение уровня p.R269C и p.R315W. Напротив, p.T701I демонстрировал уровни экспрессии белка, сходные с уровнями TRPV4 дикого типа (фиг. 3A-B). Поскольку в предыдущих исследованиях сообщалось о цитотоксических эффектах мутантных белков TRPV4, анализ цитотоксичности проводили на клетках HEK293T, трансфицированных контрольными и мутантными плазмидами.
R315W TRPV4 анализировали параллельно. Кроме того, сообщалось, что мутация p.T701I является причиной невропатии. p.T701I локализован в трансмембранном домене спирали S6 (рис. 2), рядом с сайтом связывания ваниллоида в гомологичном ионном канале TRPV1. 12 В настоящее время нет функциональных исследований, устанавливающих патогенность этого варианта при заболеваниях человека или функции белка TRPV4. Поэтому мы исследовали влияние p.S94L, p.R269C, p.R314W и p.T701I на стабильность и субклеточную локализацию TRPV4. Чтобы исследовать влияние различных вариантов на стабильность белка, TRPV4-WT и мутантные конструкции были временно экспрессированы в HEK29.3T-клетки и их экспрессию анализировали с помощью вестерн-блоттинга (фиг. 3). Анализ белка с помощью вестерн-блоттинга показал снижение уровня белка p.S94L, а также снижение уровня p.R269C и p.R315W. Напротив, p.T701I демонстрировал уровни экспрессии белка, сходные с уровнями TRPV4 дикого типа (фиг. 3A-B). Поскольку в предыдущих исследованиях сообщалось о цитотоксических эффектах мутантных белков TRPV4, анализ цитотоксичности проводили на клетках HEK293T, трансфицированных контрольными и мутантными плазмидами. Количественная оценка показала значительно более высокие уровни гибели клеток в p.S9.4L, трансфицированных клетками, по сравнению с клетками, трансфицированными TRPV4 дикого типа. Точно так же p.R269C и p.R315W также приводили к повышенной цитотоксичности. Интересно, что в клетках, трансфицированных p.T701I, не наблюдалось изменений цитотоксичности по сравнению с клетками, трансфицированными TRPV дикого типа. Клетки HEK293T, трансфицированные контрольными и мутантными плазмидами, также культивировали в присутствии антагониста канала TRPV4 (HC-067047), который обращал эффекты цитотоксичности, наблюдаемые в p. Трансфицированные клетки S94L, p.R269C и p.315W (фиг. 3C). Присутствие HC-067047 также приводило к повышенной экспрессии p.S9.4L, p.R269C и p.R315W, однако изменений экспрессии белка p.701I обнаружено не было (рис. 3B). Для изучения субклеточной локализации p.S94L и других мутантов был проведен анализ иммунофлуоресценции (рис. 4). ). TRPV4 дикого типа преимущественно локализуется на плазматической мембране, как это было описано для эндогенного белка TRPV4.
Количественная оценка показала значительно более высокие уровни гибели клеток в p.S9.4L, трансфицированных клетками, по сравнению с клетками, трансфицированными TRPV4 дикого типа. Точно так же p.R269C и p.R315W также приводили к повышенной цитотоксичности. Интересно, что в клетках, трансфицированных p.T701I, не наблюдалось изменений цитотоксичности по сравнению с клетками, трансфицированными TRPV дикого типа. Клетки HEK293T, трансфицированные контрольными и мутантными плазмидами, также культивировали в присутствии антагониста канала TRPV4 (HC-067047), который обращал эффекты цитотоксичности, наблюдаемые в p. Трансфицированные клетки S94L, p.R269C и p.315W (фиг. 3C). Присутствие HC-067047 также приводило к повышенной экспрессии p.S9.4L, p.R269C и p.R315W, однако изменений экспрессии белка p.701I обнаружено не было (рис. 3B). Для изучения субклеточной локализации p.S94L и других мутантов был проведен анализ иммунофлуоресценции (рис. 4). ). TRPV4 дикого типа преимущественно локализуется на плазматической мембране, как это было описано для эндогенного белка TRPV4. p.S94L и ранее описанные p.R269C и p.R315W проявляли локализацию на плазматической мембране. Интересно, что мутантный белок p.T701I имеет перинуклеарную локализацию, и, в отличие от белка дикого типа, белок не обнаруживается на плазматической мембране (рис. 4).
p.S94L и ранее описанные p.R269C и p.R315W проявляли локализацию на плазматической мембране. Интересно, что мутантный белок p.T701I имеет перинуклеарную локализацию, и, в отличие от белка дикого типа, белок не обнаруживается на плазматической мембране (рис. 4).
biorxiv;402388v2/FIG3F3fig3Рисунок 3:
(A-B) Вестерн-блоттинг клеток HEK293T, транзиентно экспрессирующих TRPV4-FLAG, p.S94L-FLAG, p.R269C-FLAG, p.R315W-FLAG, p.T701I-FLAG или пустой вектор собирали через 48 часов после трансфекции и инкубации в отсутствие или в присутствии HC-067047 (5 мкМ). Клеточные лизаты исследовали антителами против метки FLAG для обнаружения трансфицированных белков TRPV4 дикого типа и мутантных белков, а также GAPDH в качестве контроля загрузки. (C) Анализ цитотоксичности в клетках, трансфицированных контрольными или мутантными плазмидами TRPV4. Различия в уровнях белка или цитотоксичности считались значимыми при * p<0,05.
biorxiv;402388v2/FIG4F4fig4Рисунок 4:
Репрезентативные изображения клеток HEK293T, временно экспрессирующих белки TRPV4-Flag, p. S94L-Flag, p.R269C-Flag, p.R315W-Flag, p.T701I. Иммунофлуоресценцию проводили с антителом против флаговой метки для обнаружения белков TRPV4-Flag и Na-K-АТФазой для мечения плазматической мембраны. Ядра окрашивали DAPI. Масштабная линейка = 20 мкМ.
S94L-Flag, p.R269C-Flag, p.R315W-Flag, p.T701I. Иммунофлуоресценцию проводили с антителом против флаговой метки для обнаружения белков TRPV4-Flag и Na-K-АТФазой для мечения плазматической мембраны. Ядра окрашивали DAPI. Масштабная линейка = 20 мкМ.
Обсуждение
Дистальные наследственные моторные невропатии (дНМН) представляют собой генетически гетерогенную группу заболеваний, характеризующихся слабостью дистальных отделов нижних двигательных нейронов. Это контрастирует с болезнью ШМТ и наследственными сенсорными нейропатиями, при которых сенсорное поражение является основным компонентом заболевания. Многие формы dHMN могут демонстрировать второстепенный сенсорный компонент, и существует перекрытие между аксональными формами CMT (CMT2) и dHMN. Аутосомно-доминантные мутации в TRPV4 связаны с разнообразными клиническими проявлениями, включая болезнь dHMN, SPSMA, CSMAA и аутосомно-доминантную аксональную ШМТ типа 2C (CMT2C). Мутации TRPV4 также приводят к нескольким формам аутосомно-доминантной скелетной дисплазии. У пробанда был обнаружен артрогриопсис, связанный со слабостью дистальных мышц с нормальной сенсорной реакцией, и, следовательно, клинически диагностированный как врожденная спинальная мышечная атрофия и артрогрипоз (CSMAA), подтип dHMN. Несколько функциональных исследований продемонстрировали механизм усиления функции при мутантных TRPV4-опосредованных нейропатиях 11 . Мутантные белки TRPV4 часто обнаруживают нормальную локализацию; однако они демонстрируют повышенную активность кальциевых каналов как на базальном, так и на активированном уровне. Это приводит к увеличению внутриклеточной концентрации кальция, что приводит к цитотоксичности.
У пробанда был обнаружен артрогриопсис, связанный со слабостью дистальных мышц с нормальной сенсорной реакцией, и, следовательно, клинически диагностированный как врожденная спинальная мышечная атрофия и артрогрипоз (CSMAA), подтип dHMN. Несколько функциональных исследований продемонстрировали механизм усиления функции при мутантных TRPV4-опосредованных нейропатиях 11 . Мутантные белки TRPV4 часто обнаруживают нормальную локализацию; однако они демонстрируют повышенную активность кальциевых каналов как на базальном, так и на активированном уровне. Это приводит к увеличению внутриклеточной концентрации кальция, что приводит к цитотоксичности.
После секвенирования полного экзома и функционального анализа мы определили гомозиготный миссенс-вариант p.S94L в TRPV4 как причину тяжелой формы дистальной наследственной моторной нейропатии. Этот сериновый остаток высоко консервативен у млекопитающих, и только 6 гетерозиготных и ни одного гомозиготного человека в контрольной популяции из 246 210 человек (база данных агрегации геномов, gnomAD) сообщается только о 6 гетерозиготных и ни о каких гомозиготных особях (база данных агрегации геномов, gnomAD), что указывает на то, что это ключевая аминокислота. Здесь мы демонстрируем, что TRPV4 p.S9Вариант 4L приводил к повышенной цитотоксичности, как сообщалось ранее для других патогенных вариантов TRPV4, вызывающих невропатию. Ограничением этой работы является то, что эти исследования проводились на клетках HEK293T, а не на клетках нейронов. Однако клетки HEK293T обычно использовались для определения аберрантной функции канала для доминантных мутаций TRPV4, что позволяет сравнить наши результаты с предыдущей работой 3 . Кроме того, мутация p.S94L приводила к уменьшению количества мутантного белка, который был спасен антагонистом канала TRPV4, как ранее наблюдалось в нейрональных клетках 11 . Предыдущая работа также выявила сниженную стабильность многих доминантных вариантов во внутриклеточном домене анкириновых повторов TRPV4 13 . Недавнее исследование также идентифицировало биаллельные гетерозиготные мутации, расположенные на С-конце канала TRPV4 у двух братьев и сестер с невропатией, связанной с тяжелой умственной отсталостью 14 .
Здесь мы демонстрируем, что TRPV4 p.S9Вариант 4L приводил к повышенной цитотоксичности, как сообщалось ранее для других патогенных вариантов TRPV4, вызывающих невропатию. Ограничением этой работы является то, что эти исследования проводились на клетках HEK293T, а не на клетках нейронов. Однако клетки HEK293T обычно использовались для определения аберрантной функции канала для доминантных мутаций TRPV4, что позволяет сравнить наши результаты с предыдущей работой 3 . Кроме того, мутация p.S94L приводила к уменьшению количества мутантного белка, который был спасен антагонистом канала TRPV4, как ранее наблюдалось в нейрональных клетках 11 . Предыдущая работа также выявила сниженную стабильность многих доминантных вариантов во внутриклеточном домене анкириновых повторов TRPV4 13 . Недавнее исследование также идентифицировало биаллельные гетерозиготные мутации, расположенные на С-конце канала TRPV4 у двух братьев и сестер с невропатией, связанной с тяжелой умственной отсталостью 14 . Эти мутантные белки продемонстрировали нормальную мембранную локализацию; однако они показали снижение функции канала. Тяжелый фенотип у пробанда согласуется с наблюдаемой цитотоксичностью p.S9.4L в ячейках HEK293T.
Эти мутантные белки продемонстрировали нормальную мембранную локализацию; однако они показали снижение функции канала. Тяжелый фенотип у пробанда согласуется с наблюдаемой цитотоксичностью p.S9.4L в ячейках HEK293T.
Мутация p.S94L расположена в N-концевой внутриклеточной области перед доменом анкириновых повторов. Предыдущие исследования показали, что доминантные мутации в этой области приводят к невропатии у больных 15 . По сравнению с ранее известными вариантами TRPV4, такими как p.R269C и p.R315W, в трансфицированных клетках HEK293T были обнаружены более высокие уровни белка p.S94L TRPV4 (рис. 3 AB). Точно так же мы наблюдали более низкий уровень цитотоксичности в p.S9.4L трансфицированных клеток по сравнению с трансфицированными клетками p.R269C и p.R315W (фиг. 3C). Кроме того, тот факт, что родители пробанда, гетерозиготные по варианту TRPV4 p.S94L, не проявляли никакого фенотипа и что в базе данных gnomAD сообщается о шести гетерозиготных индивидуумах, предполагают сниженную пенетрантность этого варианта в гетерозиготном состоянии.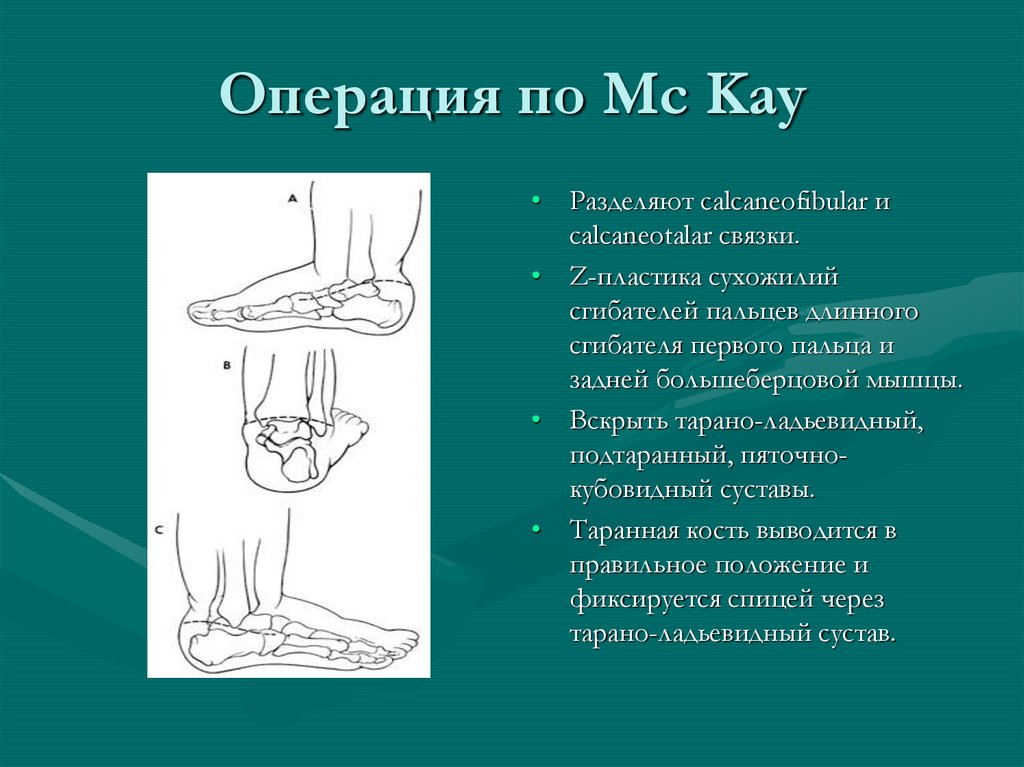 Тем не менее, многие гетерозиготные варианты TRPV4 демонстрируют сниженную пенетрантность у взрослых носителей, и по этой причине родителям пробанда было рекомендовано регулярно наблюдаться в неврологической клинике. Интересно, что большинство болезнетворных вариантов человека, изученных в этой работе, проявляли пониженную стабильность белка, тогда как вариант p.T701I изменял клеточную локализацию белка TRPV4 от плазматической мембраны до перинуклеарной области. Эта неправильная локализация может привести к изменению функции кальциевых каналов у пострадавших пациентов. Вместе эти результаты демонстрируют, что разные мутации в TRPV4 влияют на функцию канала с помощью разных механизмов. Понимание функциональных изменений в мутантных каналах TRPV4 необходимо не только для определения патогенности новых вариантов, но и для разработки специфической терапии, поскольку доступны как агонисты, так и антагонисты каналов TRPV4.
Тем не менее, многие гетерозиготные варианты TRPV4 демонстрируют сниженную пенетрантность у взрослых носителей, и по этой причине родителям пробанда было рекомендовано регулярно наблюдаться в неврологической клинике. Интересно, что большинство болезнетворных вариантов человека, изученных в этой работе, проявляли пониженную стабильность белка, тогда как вариант p.T701I изменял клеточную локализацию белка TRPV4 от плазматической мембраны до перинуклеарной области. Эта неправильная локализация может привести к изменению функции кальциевых каналов у пострадавших пациентов. Вместе эти результаты демонстрируют, что разные мутации в TRPV4 влияют на функцию канала с помощью разных механизмов. Понимание функциональных изменений в мутантных каналах TRPV4 необходимо не только для определения патогенности новых вариантов, но и для разработки специфической терапии, поскольку доступны как агонисты, так и антагонисты каналов TRPV4.
Вспомогательная информацияDC1Доступность данных
Доступ к исследовательским материалам, поддерживающим эту публикацию, можно получить, обратившись к соответствующему автору по адресу «vgupta@research. bwh.harvard.edu».
bwh.harvard.edu».
Благодарность
Эта работа была поддержана K01 AR062601 (VAG) и стипендией Элеоноры, Майлза Шора для ученых в области медицины, а также премией Brigham and Women’s Hospital Career Development Award (VAG), Премией директора Института биомедицинских исследований имени Бригама (Brigham Genomic Medicine), и Американской кардиологической ассоциации 16GRNT27250119.(РГ).
Приложение Ibiorxiv;402388v2/UTBL1T1utbl1References1.RossorAM, PolkeJM, HouldenH, ReillyMM. Клинические последствия генетических достижений при болезни Шарко-Мари-Тута. Nat Rev Neurol2013;9:562–571.2.PareysonD, SaveriP, PisciottaC.Новые разработки в области невропатии Шарко-Мари-Тута и родственных заболеваний. Curr Opin Neurol, 2017; 30:471–480.3. LandoureG, ZdebikAA, MartinezTL и др. Мутации в TRPV4 вызывают болезнь Шарко-Мари-Тута типа 2C. Nat Genet2010; 42:170–174.4.RockMJ, PrenenJ, FunariVA и др. Мутации с приобретением функции в TRPV4 вызывают аутосомно-доминантную брахиолмию. Нат Жене2008;40:999–1003. 5.HaghighiA, CassaCA, KrierJB и др. Комплексная клиническая программа и стратегия краудсорсинга для секвенирования генома и открытия генов менделевской болезни. Genomic Medicine2018.6.DengZ, PaknejadN, MaxaevG, et al. Крио-ЭМ и рентгеновские структуры TRPV4 раскрывают понимание механизмов проникновения ионов и ворот. Nat Struct Mol Biol2018;25:252–260.7.Garcia-EliasA, MrkonjicS, Pardo-PastorC, et al. Зависимая от фосфатидилинозитол-4,5-бифосфата перегруппировка цитозольных хвостов TRPV4 обеспечивает активацию канала физиологическими стимулами. Proc Natl Acad Sci U S A2013; 110:9553–9558.8. GoretzkiB, GlogowskiNA, DiehlE, et al. Структурная основа взаимодействия N-конца TRPV4 с Syndapin/PACSIN1-3 и PIP2. Структура (Лондон, Англия: 1993) 2018.9.CuajungcoMP, GrimmC, OshimaK, et al. PACSIN связываются с катионным каналом TRPV4. PACSIN 3 модулирует субклеточную локализацию TRPV4. J Biol Chem2006; 281:18753–18762.10.D’HoedtD, OwsianikG, PrenenJ, et al. Стимул-специфическая модуляция катионного канала TRPV4 с помощью PACSIN 3.
5.HaghighiA, CassaCA, KrierJB и др. Комплексная клиническая программа и стратегия краудсорсинга для секвенирования генома и открытия генов менделевской болезни. Genomic Medicine2018.6.DengZ, PaknejadN, MaxaevG, et al. Крио-ЭМ и рентгеновские структуры TRPV4 раскрывают понимание механизмов проникновения ионов и ворот. Nat Struct Mol Biol2018;25:252–260.7.Garcia-EliasA, MrkonjicS, Pardo-PastorC, et al. Зависимая от фосфатидилинозитол-4,5-бифосфата перегруппировка цитозольных хвостов TRPV4 обеспечивает активацию канала физиологическими стимулами. Proc Natl Acad Sci U S A2013; 110:9553–9558.8. GoretzkiB, GlogowskiNA, DiehlE, et al. Структурная основа взаимодействия N-конца TRPV4 с Syndapin/PACSIN1-3 и PIP2. Структура (Лондон, Англия: 1993) 2018.9.CuajungcoMP, GrimmC, OshimaK, et al. PACSIN связываются с катионным каналом TRPV4. PACSIN 3 модулирует субклеточную локализацию TRPV4. J Biol Chem2006; 281:18753–18762.10.D’HoedtD, OwsianikG, PrenenJ, et al. Стимул-специфическая модуляция катионного канала TRPV4 с помощью PACSIN 3. J Biol Chem2008;283:6272–6280.11.FectoF, ShiY, HudaR, MartinaM, SiddiqueT, DengHX. Токсичность, опосредованная мутантным TRPV4, связана с усилением конститутивной функции при аксональных нейропатиях. J Biol Chem 2011; 286: 17281–17291.12. Структуры GaoY, CaoE, JuliusD, ChengY.TRPV1 в нанодисках раскрывают механизмы действия лигандов и липидов. Nature 2016; 534: 347–351.13. InadaH, ProckoE, SotomayorM, GaudetR. Структурные и биохимические последствия болезнетворных мутаций в домене анкириновых повторов канала TRPV4 человека. Biochemistry2012;51:6195–6206.14.ThibodeauML, PetersCH, TownsendKN, et al. Сложные гетерозиготные мутации TRPV4 у двух братьев и сестер со сложным фенотипом, включая тяжелую умственную отсталость и невропатию. Am J Med Genet A2017; 173: 3087–3092.15. Мутации FiorilloC, MoroF, BriscaG и др. TRPV4 у детей с врожденной дистальной спинальной мышечной атрофией. Neurogenetics2012;13:195–203.16.BennettAH, O’DonohueMF, GundrySR и др. РНК-хеликаза DDX27 регулирует рост и регенерацию скелетных мышц путем модуляции трансляционных процессов.
J Biol Chem2008;283:6272–6280.11.FectoF, ShiY, HudaR, MartinaM, SiddiqueT, DengHX. Токсичность, опосредованная мутантным TRPV4, связана с усилением конститутивной функции при аксональных нейропатиях. J Biol Chem 2011; 286: 17281–17291.12. Структуры GaoY, CaoE, JuliusD, ChengY.TRPV1 в нанодисках раскрывают механизмы действия лигандов и липидов. Nature 2016; 534: 347–351.13. InadaH, ProckoE, SotomayorM, GaudetR. Структурные и биохимические последствия болезнетворных мутаций в домене анкириновых повторов канала TRPV4 человека. Biochemistry2012;51:6195–6206.14.ThibodeauML, PetersCH, TownsendKN, et al. Сложные гетерозиготные мутации TRPV4 у двух братьев и сестер со сложным фенотипом, включая тяжелую умственную отсталость и невропатию. Am J Med Genet A2017; 173: 3087–3092.15. Мутации FiorilloC, MoroF, BriscaG и др. TRPV4 у детей с врожденной дистальной спинальной мышечной атрофией. Neurogenetics2012;13:195–203.16.BennettAH, O’DonohueMF, GundrySR и др. РНК-хеликаза DDX27 регулирует рост и регенерацию скелетных мышц путем модуляции трансляционных процессов.