
Первичный иммунодефицит у детей что это такое: Что такое первичный иммунодефицит? / Фонд Подсолнух

Что такое первичный иммунодефицит? / Фонд Подсолнух
На дворе 21 век. Человек – царь природы. Достижения науки позволяют контролировать течение таких сложных болезней, которые раньше и диагностировать было невозможно. Лекарства открывают и внедряют в практику очень быстро. На этом фоне кажется, что человек при правильном подходе может вообще не болеть, ведь у нас расписание на день, неделю, год. В них нет времени для болезней. И тогда нарушенные планы, сорванные тренировки, занятия, встречи, переговоры заставляют нас сначала грустить, а затем и паниковать – что не так со мной или моим ребенком? Почему я или он все время болеем? Может это проявления иммунодефицита? И что такое Иммунодефицит? Когда можно не волноваться, а когда бежать к врачу за направлением на обследование? Только ли инфекционные осложнения могут навести на мысль о проблемах с иммунной системой? Может ли быть иммунодефицит вообще без инфекций?
Узнать больше про ПИД на портале — PROPID.ru
Итак, первичное иммунодефицитное состояние (ПИДС)
Конечно, заболеть может абсолютно любой человек любого возраста. Но вопрос в том, чем болеет и как переносит он эту болезнь.И, конечно, ребенок переносящий ОРЗ в детском саду в виде насморка и кашля, без осложнений, даже до 8-10 раз в год не будет иметь повода заподозрить у него ПИДС. Первичные иммунодефицитные состояния — это большая группа врожденных заболеваний, которые развиваются вследствие генетических нарушений и приводят к развитию тяжелых хронических инфекций и воспалительному поражению органов и тканей. Клинически это может проявляться не только инфекционными процессам, но и различными аутоиммунными и онкологическими заболеваниями, а так же хроническим воспалением. Согласно классификации Международного союза иммунологических обществ (IUIS) выделяют 9 групп ПИДС по механизму заболевания, которые в свою очередь подразделяются на самостоятельные единицы, с указанием пораженного гена. И, таким образом, в настоящее время открыто более 400 вариантов ПИДС.
Но вопрос в том, чем болеет и как переносит он эту болезнь.И, конечно, ребенок переносящий ОРЗ в детском саду в виде насморка и кашля, без осложнений, даже до 8-10 раз в год не будет иметь повода заподозрить у него ПИДС. Первичные иммунодефицитные состояния — это большая группа врожденных заболеваний, которые развиваются вследствие генетических нарушений и приводят к развитию тяжелых хронических инфекций и воспалительному поражению органов и тканей. Клинически это может проявляться не только инфекционными процессам, но и различными аутоиммунными и онкологическими заболеваниями, а так же хроническим воспалением. Согласно классификации Международного союза иммунологических обществ (IUIS) выделяют 9 групп ПИДС по механизму заболевания, которые в свою очередь подразделяются на самостоятельные единицы, с указанием пораженного гена. И, таким образом, в настоящее время открыто более 400 вариантов ПИДС.
Исторически считалось, что ПИДС – крайне редкое заболевание. Однако по мере накопления знаний, это утверждение требует уточнения. Предполагаемая частота встречаемости ПИДС в мире очень сильно меняется в зависимости от социальных и национальных особенностей, точности клинико-лабораторных критериев постановки диагноза, методов сбора данных. Во всем мире единственным способом для сбора и накопления информации по редким заболеваниям являются так называемые регистры. Они могут быть национальные отдельных стран, а могут быть объединены в более крупные, что позволяет расширить знания по самым редко встречающимся состояниям. Так регистр Европейского общества ПИДС существует с 2004 г. и содержит данные о более чем 28 тыс. пациентов. В Российской федерации с помощью Национальной Ассоциации экспертов в области ПИД (НАЭПИД) в 2017 г. создан российский регистр, в котором к настоящему времени внесены данные около 3.5тыс пациентов. Но, если учитывать средние показатели распространенности ПИДС в мире, то в России таких пациентов должно было бы быть не менее 12-14 тыс.
Однако по мере накопления знаний, это утверждение требует уточнения. Предполагаемая частота встречаемости ПИДС в мире очень сильно меняется в зависимости от социальных и национальных особенностей, точности клинико-лабораторных критериев постановки диагноза, методов сбора данных. Во всем мире единственным способом для сбора и накопления информации по редким заболеваниям являются так называемые регистры. Они могут быть национальные отдельных стран, а могут быть объединены в более крупные, что позволяет расширить знания по самым редко встречающимся состояниям. Так регистр Европейского общества ПИДС существует с 2004 г. и содержит данные о более чем 28 тыс. пациентов. В Российской федерации с помощью Национальной Ассоциации экспертов в области ПИД (НАЭПИД) в 2017 г. создан российский регистр, в котором к настоящему времени внесены данные около 3.5тыс пациентов. Но, если учитывать средние показатели распространенности ПИДС в мире, то в России таких пациентов должно было бы быть не менее 12-14 тыс. Таким образом, большинство пациентов все-таки остаются не диагностированы.
Таким образом, большинство пациентов все-таки остаются не диагностированы.
Так как же заподозрить этот сложный диагноз?
Первичные признаки врожденного иммунодефицита могут быть не обнаружены, потому что у него нет уникальных черт. Он может проявляться как «обычная повторяющаяся инфекция», например носа, ушей или легких. Могут возникать желудочно-кишечные проблемы или воспаление суставов. Родители и врачи часто не осознают, что эти проблемы созданы генетическими дефектами иммунной системы. Инфекции переходят в хроническую форму, создают осложнения. Тяжелые формы первичного иммунодефицита обычно проявляются сразу после рождения ребенка или спустя некоторое время после него.
Это именно частые бактериальные инфекции, например – 8 и более гнойных отитов или больше 2 тяжелыхгнойных синуситов или осложненных пневмоний в год. Это устойчивость инфекционного процесса к антибактериальной терапии, из-за чего лечение многими курсами современных антибиотиков может проводиться более 2 месяцев. В эту же группу подозрений должны войти больные, перенесшие две и более генерализованных инфекции (менингит, остеомиелит, сепсис) или рецидивирующие глубокие абсцессы кожи и мягких тканей, а так же имеющие осложнения после вакцинации ослабленными живыми вакцинами (БЦЖ, полиомиелит).
В эту же группу подозрений должны войти больные, перенесшие две и более генерализованных инфекции (менингит, остеомиелит, сепсис) или рецидивирующие глубокие абсцессы кожи и мягких тканей, а так же имеющие осложнения после вакцинации ослабленными живыми вакцинами (БЦЖ, полиомиелит).
Но все же патология иммунной системы гораздо более широкое понятие, чем просто отсутствие противоинфекционной защиты. Могут быть нарушены такие функции, как защитное воспаление, и тогда это проявится воспалением неконтролируемым. Такие больные могут иметь клинику тяжелого дерматита, рецидивирующей крапивницы, отеков, устойчивых к антигистаминной и даже гормональной терапии. Должны насторожить эпизоды периодически возникающей лихорадки, увеличенных лимфоузлов, которые сопровождаются воспалительными изменениями в лабораторных анализах, но при этом не имеют признаков наличия инфекционного процесса. Гипервоспаление может коснуться кишечника, и тогда при его раннем начале это будет приводить к дефициту массы тела.
Учитывая имеющуюся в настоящее время возможность проведения различных генетических исследований для постановки многих диагнозов, необходимо использовать ее как можно раньше, т.к. по статистике самая высокая смертность от осложнений при ПИДС приходится на 1 год жизни и ранний возраст. Поэтому ранняя постановка диагноза и своевременно оказанное лечение может спасти много жизней и существенно уменьшить количество тяжелых инвалидизирующих последствий неправильно леченных больных.
Исследование Brown показало, что выживаемость после трансплантации гемопоэтических стволовых клеток (ТГСК) у пациентов с самым тяжелым иммунодефицитом ТКИН (тяжелая комбинированная иммунная недостаточность), диагностированых в первые недели жизни , составила 92 %, а при постановке диагноза в среднем к 143 дням жизни лишь 61%. В связи с этим уже сейчас идет подготовка к внедрению неонатального скрининга на ПИДС (обследование всех новорожденных в роддоме). Так же нужно обращать внимание на такие диагностируемые после рождения состояния, как отсутствие или уменьшение тимуса (вилочковой железы), миндалин, микроцефалия (маленький размер головы), расщелины твердого и мягкого неба («заячья губа», «волчья пасть»), т. к. эти состояния могут сочетаться с патологией иммунной системы. Такие дети должны как можно раньше попасть на консультацию к иммунологу.
к. эти состояния могут сочетаться с патологией иммунной системы. Такие дети должны как можно раньше попасть на консультацию к иммунологу.
Но повышенная настороженность по отношению к ПИДС должна быть не только у педиатров. В регистре ПИДС 1/3 пациентов представлена взрослыми. Нестандартное течение инфекционного процесса или аутоиммунного заболевания, устойчивость к проводимой врачами терапии, должны являться поводом для более глубокого обследования такого больного врачом- иммунологом.
Прогноз заболевания зависит от времени постановки правильного диагноза и начатого лечения.
Отдельно хотелось бы отметить низкую осведомленность в нашей стране населения и даже врачей о возможности диагностики генетических заболеваний еще до рождения ребенка, если в семье уже есть больной с установленным диагнозом ПИДС. Это помогает подготовиться к рождению такого ребенка и максимально быстро оказать необходимую помощь и лечение, позволяющее избежать жизнеугрожающих осложнений.
Пренатальная диагностика и генетическое консультирование.

На сегодняшний день установлено, что многие иммунодефициты являются наследственными заболеваниями: известен тип наследования, выявлена локализация дефектного гена, определен продукт этого гена. Cтановится ясно, что первичный иммунодефицит – не столь редкое состояние, как принято было считать. Родителям и родственникам детей, больных ПИД необходимо знать, что в настоящее время стало возможным выявление носительства дефектного гена и генетическое консультирование семей, планирующих рождение детей с риском возникновения ПИД. Например, выявление одного из самых страшных диагнозов ПИД Тяжелой Комбинированной Иммунной Недостаточности (диагноза, являющегося 100 % летальным без проведения трансплантации костного мозга) возможно до рождения ребенка, если в семье уже есть больной ребенок, а также, если выявлен молекулярный дефект. Если у ранее рожденного больного ребенка проведен анализ мутации можно выполнить диагностическое исследование нерожденного ребенка ( эмбриона или плода с околоплодными тканями). Это можно сделать путем молекулярного анализа ресничек хориона или околоплодной жидкости, содержащей клетки плода и полученной из полости матки путем амниоцентеза.
Это можно сделать путем молекулярного анализа ресничек хориона или околоплодной жидкости, содержащей клетки плода и полученной из полости матки путем амниоцентеза.
В России проведение пренатальной диагностики и генетическое консультирование возможно в следующих медицинских диагностических центрах:
— Медико-генетический научный центр Российской академии медицинских наук (МГНЦ РАМН) http://www.med-gen.ru/
Адрес : 115478, Москва, ул. Москворечье, д. 1, Медико-генетический научный центр РАМН, второй этаж ; проезд: метро «Каширская»
Телефоны регистратуры: (495) 324-87-72, 324-18-65, 324-31-57
Консультативный прием осуществляется с 10.00 до 15.00 ежедневно, кроме субботы и воскресенья по предварительной записи по телефонам: (495) 324-87-72, 324-18-65, 324-31-57
— «Центр Молекулярной Генетики» Полякова ; http://www.dnalab.ru/
115478, Москва, ул. Москворечье, д. 1, в здании Медико-генетического научного центра на 1ом этаже, каб. 116; проезд: метро «Каширская»
116; проезд: метро «Каширская»
Телефоны регистратуры: (495) 221-90-84, (495) 324-05-01, (495) 324-81-10, (495) 324-98-46
Часто задаваемые вопросы по первичному иммунодефициту
Первичный иммунодефицит – это СПИД?
Нет, первичные иммунодефициты являются наследственными заболевания и не относятся к инфекционным заболеваниям, т.к. дети страдающие первичным иммунодефицитом не опасны для окружающих, скорее окружающие представляют угрозу для детей страдающих первичными иммунодефицита.
Можно ли вылечить первичный иммунодефицит?
На сегодняшний день существует возможность излечения от первичного иммунодефицита, с помощью трансплантации костного мозга. В настоящее время в мире в каждым днем увеличивается количество форм первичных иммунодефицитов, которые можно вылечить с помощью ТКМ. Если еще несколько лет назад считалось что достаточно назначить антибактериальную и заместительную терапию иммуноглобулином, но на сегодняшний день во всем мире стараются как можно раньше провести трансплантацию костного мозга, чтобы избежать различных осложнений.
А не страшно ли то, что больные первичными иммунодефицитами всю жизнь получают антибиотики и другие противомикробные препараты, если считается что антибиотики подавляют иммунитет?
При первичных иммунодефицитах антибактериальная и другая противомикробная терапия назначается для помощи организму т.к. в первую очередь страдает противоинфекционная защита. В большинстве случаев, врачи, занимающими лечением пациентов, страдающих первичными иммунодефицитами, не видят осложнений от антибактериальной терапии, о которых так широко говориться в различных средствах массовой информации.
Если в семье есть дети, страдающие первичным иммунодефицитом, это значит, что у этой семьи не может здоровых детей?
Вероятность рождения больного ребенка составляет 25% независимо от варианта наследования заболевания. В настоящее время известны гены, мутации в которых приводят к развитию первичных иммунодефицитов. В большинстве случаев врачи способны помочь семье, проведя на ранних сроках беременности пренатальную диагностику и установив страдает ли плод первичным иммунодефицитом.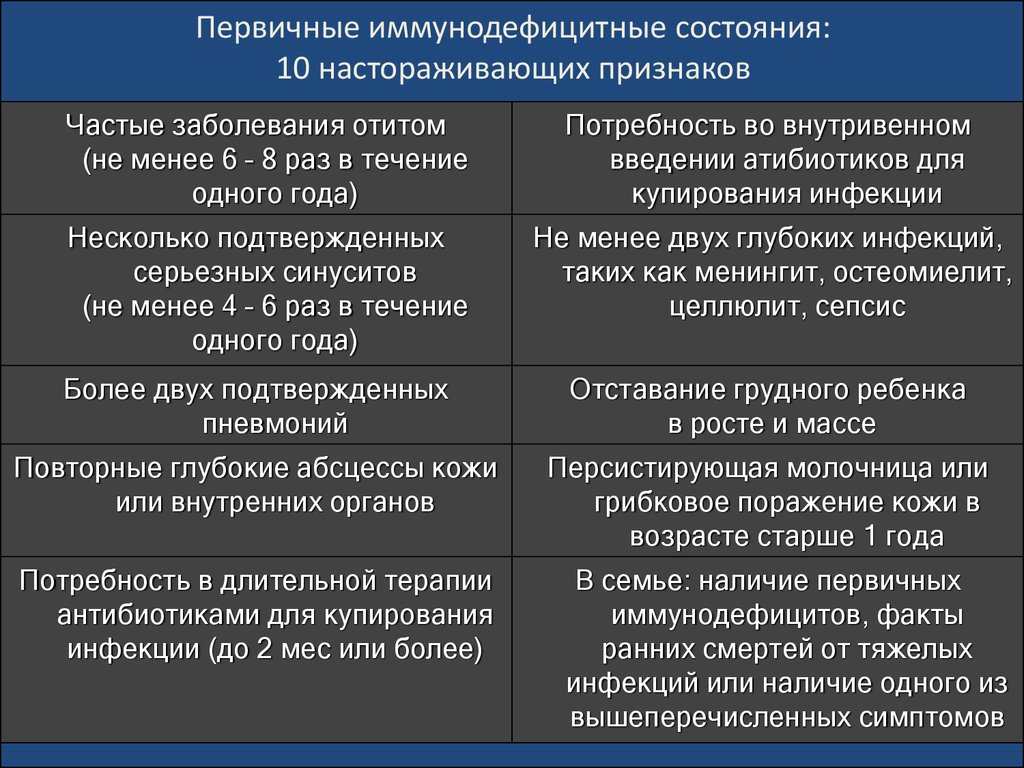
Существуют ли ограничения в социальной жизни у детей, страдающих первичными иммунодефицитами?
В большинстве случаев эти дети ведут такой же образ жизни, как и все остальные, исключения составляют дети, страдающие тяжелыми формами иммунодефицита, например ТКИН, которые нуждаются в изоляции и больные после проведения ТКМ.
Автор-составитель – Юрасова Анна, врач-иммунолог
ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ У ДЕТЕЙ: ТРЕВОЖНЫЕ ПРИЗНАКИ
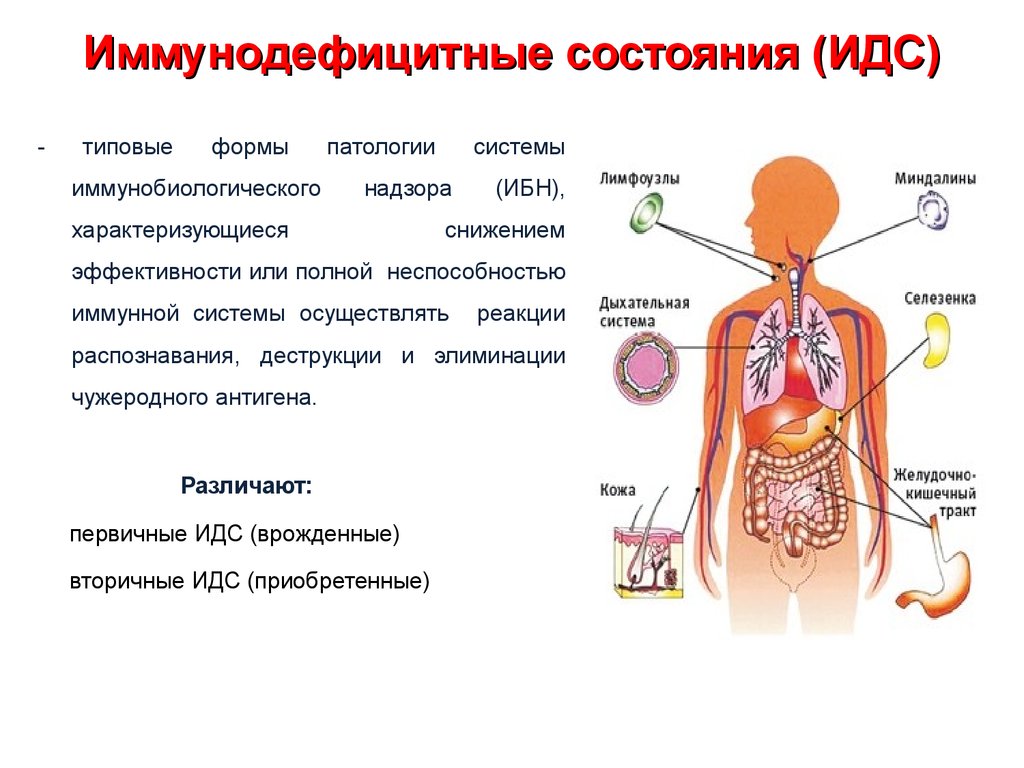
Первичный иммунодефицит (ПИД) – это состояние, при котором иммунитет человека не способен выполнять свою защитную функцию. Врожденные иммунодефицитные состояния, как правило, проявляются у детей в течение первых двух лет жизни и приводят к возникновению инфекций, которые необычайно трудно поддаются лечению, рецидивируют, быстро переходят в хроническую форму и в конечном счете являют собой угрозу для жизни ребенка.
Профильная медицинская организация, в которой проводится диагностика первичного иммунодефицита у детей в Москве, — Консультативно-диагностический центр аллергологии и иммунологии ДГКБ №9 им. Сперанского.
Сперанского.
Что нужно знать о первичном иммунодефиците?
1. Первичным иммунодефицитом нельзя заразиться, это почти всегда либо наследственное состояние, либо приобретенное во внутриутробном периоде. Ребенок с ПИД может родиться у совершенно здоровых родителей в результате случайной генной мутации.
2. Существует более 300 форм ПИД, среди которых как сравнительно легкие, так и тяжелые, опасные для жизни заболевания. С некоторыми формами ПИД пациенты могут жить до старости, при условии, что они будут получать надлежащую пожизненную терапию. В некоторых случаях специфической терапии не предусмотрено.
3. ПИД чаще всего проявляется в течение первых двух лет жизни, но при некоторых формах иммунодефицит может проявить себя и в более старшем возрасте.
4. Самое главное, что можно сделать – это вовремя диагностировать иммунодефицит. Симптомы этого состояния неспецифичны, поэтому велик риск «пропустить» тревожные сигналы. Между тем, для жизни и здоровья ребенка необходимо, чтобы он мог как можно скорее получать необходимое лечение. Российская детская клиническая больница и Институт иммунологии насчитывают около 2000 пациентов с первичным иммунодефицитом, однако детей с не диагностированным ПИД может оказаться во много раз больше.
Российская детская клиническая больница и Институт иммунологии насчитывают около 2000 пациентов с первичным иммунодефицитом, однако детей с не диагностированным ПИД может оказаться во много раз больше.
Настораживающие признаки первичного иммунодефицита
Чаще всего, если ребенок болеет респираторными заболеваниями чаще 6-7 раз в год, его родители начинают беспокоиться: не говорит ли это об иммунодефиците? Чаще всего – нет, однако есть несколько тревожных сигналов, которые могут указывать на развитие ПИД.
Врачи-иммунологи советуют внимательно присмотреться к часто болеющим детям из вашего окружения. Если у ребенка есть два или больше тревожных признаков из перечисленных ниже, это повод для срочного обращения к врачу-иммунологу на предмет выявления первичного иммунодефицита.
1. Наличие диагноза ПИД у кого-то из близких родственников, случаи ранних смертей от инфекций, не поддающихся лечению.
2. Два или более тяжелых синуситов в течение года
3. Четыре или более тяжелых отитов в течение года
4. Две или более пневмоний в течение года
Две или более пневмоний в течение года
5. Длительная антибиотикотерапия (в течение двух месяцев и более), без эффекта или с минимальным эффектом.
6. Необходимость внутривенного введения антибиотиков для лечения инфекций.
7. Две или более глубоких генерализованных инфекций (менингит, сепсис, остеомиелит, септический артрит и другие).
8. У новорожденных: неспособность нормально прибавлять в росте и весе.
9. У детей от года: стойкая молочница полости рта или грибковая инфекция кожи и слизистых.
10. Повторные глубокие абсцессы кожи или внутренних органов.
Также могут вызвать опасение неясные эритемы у детей грудного возраста, нарушения переваривания в период грудного вскармливания, осложнения при вакцинации ребенка живыми ослабленными вакцинами (полиомиелит, БЦЖ).
Куда обратиться за помощью?
Обследование детей на предмет первичного иммунодефицита в Москве можно пройти в центре аллергологии и иммунологии Детской городской клинической больницы №9 им. Сперанского. Чтобы записаться на прием, обратитесь к участковому врачу-педиатру и попросите его выписать направление на консультацию к иммунологу. Вы также можете обратиться в отдел платных услуг.
Сперанского. Чтобы записаться на прием, обратитесь к участковому врачу-педиатру и попросите его выписать направление на консультацию к иммунологу. Вы также можете обратиться в отдел платных услуг.
Специалисты Центра настоятельно рекомендуют не проводить самостоятельных обследований в частных клиниках, так как при консультации врача они могут быть бесполезны. Лучше сразу обратиться к специалисту и выстроить единую стратегию.
Консультативно-диагностический центр аллергологии и иммунологии находится по адресу:
г. Москва, Шмитовский проезд, д.29
Телефон для справок: +7 499 259-0108, +7 499 259-1307, +7 499 259-1102
http://navigator.mosgorzdrav.ru/columns/allergologiya_immunologiya/pervichnyy_immunodefitsit_u_detey_trevozhnye_priznaki/
Упрощенный подход к ребенку с первичным иммунодефицитом
1. Picard C, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, Conley ME, et al. Первичные иммунодефицитные заболевания: обновление классификации Экспертного комитета Международного союза иммунологических обществ по первичному иммунодефициту 2015 г. J Clin Immunol. 2015; 35: 696–726. [Бесплатная статья PMC] [PubMed] [Google Scholar]
J Clin Immunol. 2015; 35: 696–726. [Бесплатная статья PMC] [PubMed] [Google Scholar]
2. Cunningham-Rundles C. Аутоиммунитет при первичном иммунодефиците: уроки наших пациентов. Клин Эксп Иммунол. 2011; 164 (Приложение 2): 6–11. [Бесплатная статья PMC] [PubMed] [Google Scholar]
3. Бойл Дж.М., Бакли Р.Х. Распространенность среди населения диагностированных первичных иммунодефицитных заболеваний в США. Дж. Клин Иммунол. 2007; 27: 497–502. [PubMed] [Google Scholar]
4. Сингх С., Гупта С. Первичные иммунодефицитные заболевания: потребность в осведомленности и защите интересов в Индии. Индийский J Педиатр. 2016; 83: 328–30. [PubMed] [Google Scholar]
5. Реда С.М., Эль-Гоними Д.Х., Афифи Х.М. Клинические предикторы первичных иммунодефицитных заболеваний у детей. Аллергия Астма Immunol Res. 2013; 5: 88–95. [Бесплатная статья PMC] [PubMed] [Google Scholar]
6. Modell V, Quinn J, Orange J, Notarangelo LD, Modell F. Первичные иммунодефициты во всем мире: обновленный обзор глобальной сети центров Jeffrey Modell. Иммунол Рез. 2016; 64: 736–53. [PubMed] [Google Scholar]
Иммунол Рез. 2016; 64: 736–53. [PubMed] [Google Scholar]
7. Lee JH, Son SW, Cho SH. Всесторонний обзор лечения атопической экземы. Аллергия Астма Immunol Res. 2016; 8: 181–90. [Бесплатная статья PMC] [PubMed] [Google Scholar]
8. Thomsen SF. Атопический дерматит: естественная история, диагностика и лечение. Int Sch Res Нет. 2014; 2014: e354250. [Академия Google]
9. Сакчидананд С., Сахана М.С., Аша Г.С., Шилпа К. Характер детских дерматозов в специализированном центре. Индийский J Педиатр. 2014; 81: 375–80. [PubMed] [Google Scholar]
10. Пичард Д.С., Фримен А.Ф., Коуэн Э.В. Обновление первичного иммунодефицита: Часть I. Синдромы, связанные с экзематозным дерматитом. J Am Acad Дерматол. 2015;73:355. [Бесплатная статья PMC] [PubMed] [Google Scholar]
11. Buchbinder D, Nugent DJ, Fillipovich AH. Синдром Вискотта-Олдрича: диагностика, текущее лечение и новые методы лечения. Приложение Clin Genet. 2014;7:55–66. [Бесплатная статья PMC] [PubMed] [Google Scholar]
12. Массаад М.Дж., Рамеш Н., Геха Р.С. Синдром Вискотта-Олдрича: всесторонний обзор. Энн Н.Ю. Академия наук. 2013;1285:26–43. [PubMed] [Google Scholar]
Массаад М.Дж., Рамеш Н., Геха Р.С. Синдром Вискотта-Олдрича: всесторонний обзор. Энн Н.Ю. Академия наук. 2013;1285:26–43. [PubMed] [Google Scholar]
13. Катуччи М., Кастьелло М.С., Пала Ф., Бостикардо М., Вилла А. Аутоиммунитет при синдроме Вискотта-Олдрича: неразгаданная загадка. Фронт Иммунол. 2012;3:209. [Бесплатная статья PMC] [PubMed] [Google Scholar]
14. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Рак, связанный с иммунодефицитом: обновление и перспективы. Фронт Иммунол. 2016;7:365. [Бесплатная статья PMC] [PubMed] [Google Scholar]
15. Yong PFK, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. Обновление синдромов гипер-IgE. Артрит Res Ther. 2012;14:228. [Бесплатная статья PMC] [PubMed] [Google Scholar]
16. Минегиши Ю., Карасуяма Х. Синдром гипериммуноглобулина Е и дефицит тирозинкиназы 2. Курр Опин Аллергия Клин Иммунол. 2007; 7: 506–9. [PubMed] [Google Scholar]
17. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Дефицит тирозинкиназы 2 человека выявляет ее необходимую роль в множественных цитокиновых сигналах, участвующих во врожденном и приобретенном иммунитете. Иммунитет. 2006; 25: 745–55. [PubMed] [Академия Google]
Дефицит тирозинкиназы 2 человека выявляет ее необходимую роль в множественных цитокиновых сигналах, участвующих во врожденном и приобретенном иммунитете. Иммунитет. 2006; 25: 745–55. [PubMed] [Академия Google]
18. Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramirez-Alejo N, Kilic SS, et al. Дефицит TYK2 человека: микобактериальные и вирусные инфекции без синдрома гипер-IgE. J Эксперт Мед. 2015; 212:1641–62. [Бесплатная статья PMC] [PubMed] [Google Scholar]
19. Ян Л., Флигауф М., Гримбахер Б. Синдромы гипер-IgE: обзор дефицита PGM3. Curr Opin Педиатр. 2014; 26: 697–703. [PubMed] [Google Scholar]
20. Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Мутации аутосомно-рецессивной фосфоглюкомутазы 3 (PGM3) связывают дефекты гликозилирования с атопией, иммунодефицитом, аутоиммунитетом и нейрокогнитивными нарушениями. J Аллергия Клин Иммунол. 2014;133:1400. [Бесплатная статья PMC] [PubMed] [Google Scholar]
21. Димитрова Д, Фриман А.Ф. Текущий статус посвященного иммунодефицита, связанного с цитокинезом: DOCK8 и DOCK2. Дерматол клин. 2017;35:11–9. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Дерматол клин. 2017;35:11–9. [Бесплатная статья PMC] [PubMed] [Google Scholar]
22. Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Кожные проявления синдрома дефицита DOCK8. Арка Дерматол. 2012; 148:79–84. [Бесплатная статья PMC] [PubMed] [Google Scholar]
23. Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, et al. Расширенный клинический фенотип 64 пациентов с дефицитом выделенного цитокинеза 8. J Аллергия Клин Иммунол. 2015; 136:402–12. [Бесплатная статья PMC] [PubMed] [Google Scholar]
24. Баккетта Р., Барзаги Ф., Ронкароло М.-Г. От синдрома IPEX до мутации FOXP3: урок иммунной дисрегуляции. Энн Н.Ю. Академия наук. 2016 [Epub перед печатью] [PubMed] [Google Scholar]
25. Лоренцини Т., Дотта Л., Джакомелли М., Вайро Д., Бадолато Р. Мутации STAT как переключатели программ: превращение первичных иммунодефицитов в аутоиммунные заболевания. Дж. Лейкок Биол. 2017;101:29–38. [PubMed] [Google Scholar]
26. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al.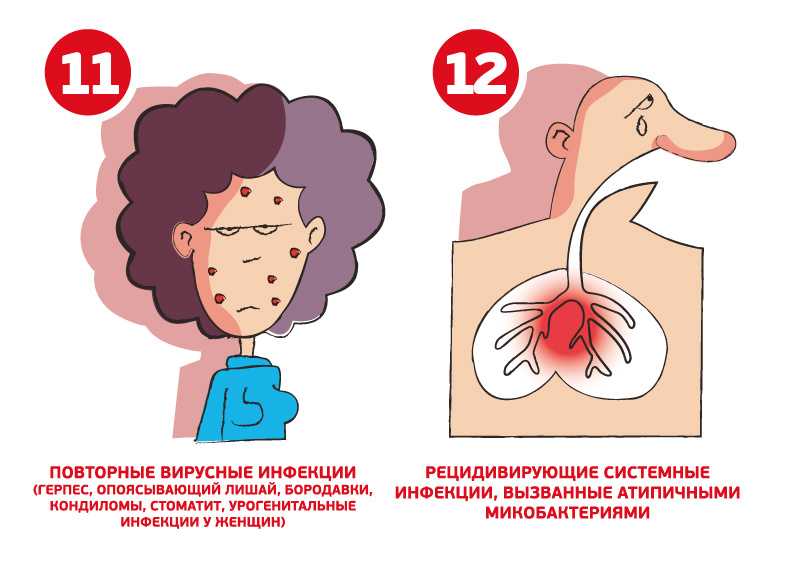 Расширенный клинический фенотип 26 пациентов с хроническим кожно-слизистым кандидозом из-за мутаций с усилением функции в STAT1. Дж. Клин Иммунол. 2016; 36:73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Расширенный клинический фенотип 26 пациентов с хроническим кожно-слизистым кандидозом из-за мутаций с усилением функции в STAT1. Дж. Клин Иммунол. 2016; 36:73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar]
27. Лейдинг Дж.В., Холланд С.М. Бородавки и все такое: вирус папилломы человека при первичных иммунодефицитах. J Аллергия Клин Иммунол. 2012; 130:1030–48. [Бесплатная статья PMC] [PubMed] [Google Scholar]
28. Диаз Г.А., Гулино А.В. Синдром WHIM: дефект передачи сигналов CXCR4. Curr Allergy Asthma Rep. 2005; 5:350–5. [PubMed] [Google Scholar]
29. Kawai T, Malech HL. Синдром WHIM: врожденный иммунодефицит. Карр Опин Гематол. 2009;16:20–6. [Бесплатная статья PMC] [PubMed] [Google Scholar]
30. Pozzobon T, Goldoni G, Viola A, Molon B. Передача сигналов CXCR4 в норме и при болезни. Иммунол Летт. 2016; 177:6–15. [PubMed] [Google Scholar]
31. Freitas C, Wittner M, Nguyen J, Rondeau V, Biajoux V, Aknin M-L, et al. Лимфоидная дифференцировка гемопоэтических стволовых клеток требует эффективной десенсибилизации Cxcr4. J Эксперт Мед. 2017 [Epub перед печатью] [Бесплатная статья PMC] [PubMed] [Google Scholar]
J Эксперт Мед. 2017 [Epub перед печатью] [Бесплатная статья PMC] [PubMed] [Google Scholar]
32. Коллин М., Дикинсон Р., Бигли В. Гематопоэтические и иммунные дефекты, связанные с мутацией GATA2. Бр Дж Гематол. 2015;169: 173–87. [Бесплатная статья PMC] [PubMed] [Google Scholar]
33. Griese M, Zarbock R, Costabel U, Hildebrandt J, Theegarten D, Albert M, et al. Дефицит GATA2 у детей и взрослых с тяжелым легочным альвеолярным протеинозом и гематологическими нарушениями. BMC Пульм Мед. 2015;15:87. [Бесплатная статья PMC] [PubMed] [Google Scholar]
34. Hsu AP, McReynolds LJ, Holland SM. Дефицит GATA2. Курр Опин Аллергия Клин Иммунол. 2015;15:104–9. [Бесплатная статья PMC] [PubMed] [Google Scholar]
35. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. Дефицит GATA2: многогранное нарушение кроветворения, лимфатической системы и иммунитета. Кровь. 2014; 123:809–21. [Бесплатная статья PMC] [PubMed] [Google Scholar]
36. Burger B, Itin PH. Бородавчатая эпидермодисплазия. Курр Пробл Дерматол. 2014;45:123–31. [PubMed] [Google Scholar]
Бородавчатая эпидермодисплазия. Курр Пробл Дерматол. 2014;45:123–31. [PubMed] [Google Scholar]
37. Artac H, Bozkurt B, Talim B, Reisli I. Саркоидноподобные гранулемы при обычном вариабельном иммунодефиците. Ревматол Интерн. 2009;30:109–12. [PubMed] [Google Scholar]
38. Агамохаммади А., Аболхассани Х., Резаи Н., Калантари Н., Тамизифар Б., Чераги Т. и др. Кожные гранулемы при общем вариабельном иммунодефиците: клинический случай и обзор литературы. Acta Dermatovenerol Croat ADC. 2010;18:107–13. [PubMed] [Google Scholar]
39. Sillevis Smitt JH, Kuijpers TW. Кожные проявления первичного иммунодефицита. Curr Opin Педиатр. 2013;25:492–7. [PubMed] [Google Scholar]
40. Geier CB, Piller A, Linder A, Sauerwein KMT, Eibl MM, Wolf HM. Негерметичный дефицит RAG у взрослых пациентов с нарушением выработки антител против бактериальных полисахаридных антигенов. PloS Один. 2015;10:e0133220. [Бесплатная статья PMC] [PubMed] [Google Scholar]
41. Пашич С., Кандольф-Секулович Л. , Джуричич С., Золотаревски Л., Симич Р., Абинун М. Некробиотические кожные гранулемы при синдроме разрыва Неймегена. J Investig Allergol Clin Immunol. 2012; 22:138–40. [PubMed] [Google Scholar]
, Джуричич С., Золотаревски Л., Симич Р., Абинун М. Некробиотические кожные гранулемы при синдроме разрыва Неймегена. J Investig Allergol Clin Immunol. 2012; 22:138–40. [PubMed] [Google Scholar]
42. Yoo J, Wolgamot G, Torgerson TR, Sidbury R. Кожные неказеозные гранулемы, связанные с синдромом разрыва Неймегена. Арка Дерматол. 2008; 144:418–9. [PubMed] [Google Scholar]
43. Aderibigbe OM, Priel DL, Lee C-CR, Ombrello MJ, Prajapati VH, Liang MG, et al. Отличительные кожные проявления и холодовая активация лейкоцитов, связанная с мутациями PLCG2. ДЖАМА Дерматол. 2015; 151: 627–34. [Бесплатная статья PMC] [PubMed] [Google Scholar]
44. Шоймер И., Райт Н., Хабер Р.М. Неинфекционные гранулемы: признак основного иммунодефицита? J Cutan Med Surg. 2016;20:259–62. [PubMed] [Google Scholar]
45. Chiam LYT, Verhagen MMM, Haraldsson A, Wulffraat N, Driessen G-J, Netea MG и соавт. Кожные гранулемы при атаксии, телеангиэктазии и других первичных иммунодефицитах: отражение неадекватной иммунной регуляции? Дерматол Базель Свитц. 2011; 223:13–9. [PubMed] [Google Scholar]
2011; 223:13–9. [PubMed] [Google Scholar]
46. Harp J, Coggshall K, Ruben BS, Ramirez-Valle F, He SY, Berger TG. Кожные гранулемы при первичном иммунодефиците: отчет о четырех случаях и обзор литературы. Int J Дерматол. 2015;54:617–25. [PubMed] [Академия Google]
47. Стегерт М., Бок М., Тренделенбург М. Клиническая картина дефицита C1q у человека: насколько волчанка? Мол Иммунол. 2015;67:3–11. [PubMed] [Google Scholar]
48. Брайан А.Р., Ву Э.Ю. Дефицит комплемента при системной красной волчанке. Curr Allergy Asthma Rep. 2014; 14:448. [PubMed] [Google Scholar]
49. Бхаттад С., Рават А., Гупта А., Сури Д., Гарг Р., де Бур М. и соавт. Дефицит раннего компонента комплемента в одноцентровой когорте педиатрической волчанки. Дж. Клин Иммунол. 2015; 35: 777–85. [PubMed] [Академия Google]
50. Macedo ACL, Исаак Л. Системная красная волчанка и дефицит ранних компонентов классического пути комплемента. Фронт Иммунол. 2016;7:55. [Бесплатная статья PMC] [PubMed] [Google Scholar]
51. Azizi G, Abolhassani H, Asgardoon MH, Alinia T, Yazdani R, Mohammadi J, et al. Аутоиммунитет при общем вариабельном иммунодефиците: эпидемиология, патофизиология и лечение. Эксперт преподобный Клин Иммунол. 2017;13:101–15. [PubMed] [Google Scholar]
Azizi G, Abolhassani H, Asgardoon MH, Alinia T, Yazdani R, Mohammadi J, et al. Аутоиммунитет при общем вариабельном иммунодефиците: эпидемиология, патофизиология и лечение. Эксперт преподобный Клин Иммунол. 2017;13:101–15. [PubMed] [Google Scholar]
52. Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Аутоиммунитет и инфекция при общем вариабельном иммунодефиците (ОВИН) Autoimmun Rev. 2016;15:877–82. [PubMed] [Академия Google]
53. Cunningham-Rundles C. Многоликий общий вариабельный иммунодефицит. Программа Hematol Educ Am Soc Hematol Am Soc Программа Hematol Educ. 2012; 2012: 301–5. [Бесплатная статья PMC] [PubMed] [Google Scholar]
54. Gualdi G, Lougaris V, Baronio M, Vitali M, Tampella G, Moratto D, et al. Бремя кожных заболеваний при селективном дефиците IgA и общем вариабельном иммунодефиците. J Investig Allergol Clin Immunol. 2015;25:369–71. [PubMed] [Google Scholar]
55. Ханна С., Этциони А. Дефицит адгезии лейкоцитов. Энн Н. Ю. Академия наук. 2012;1250:50–5. [PubMed] [Академия Google]
Ю. Академия наук. 2012;1250:50–5. [PubMed] [Академия Google]
56. Харрис Э.С., Вейрих А.С., Циммерман Г.А. Уроки редких заболеваний: синдромы дефицита адгезии лейкоцитов. Карр Опин Гематол. 2013;20:16–25. [Бесплатная статья PMC] [PubMed] [Google Scholar]
57. Madkaikar M, Italia K, Gupta M, Desai M, Aggarwal A, Singh S, et al. Дефицит адгезии лейкоцитов-I с новой интронной мутацией, проявляющейся поражениями, подобными гангренозной пиодермии. Дж. Клин Иммунол. 2015;35:431–4. [PubMed] [Google Scholar]
58. AIBarrak ZM, Alqarni AS, Chalisserry EP, Anil S. Синдром папийона-Лефевра: серия из пяти случаев среди братьев и сестер. J Med Case Rep. 2016; 10:260. [Бесплатная статья PMC] [PubMed] [Google Scholar]
59. Чаубаль Т., Бапат Р., Вадкар П. Синдром Папийона Лефевра. QJM Mon J Assoc Phys. 2017 [Epub перед печатью] [Google Scholar]
60. Dalgıc B, Bukulmez A, Sarı S. Эпоним: синдром Папийона-Лефевра. Eur J Педиатр. 2011;170:689–91. [PubMed] [Google Scholar]
61. Sebnem Kilic S, Kavurt S, Balaban Adim S. Трансфузионно-ассоциированная реакция «трансплантат против хозяина» при тяжелом комбинированном иммунодефиците. J Investig Allergol Clin Immunol. 2010;20:153–156. [PubMed] [Академия Google]
Sebnem Kilic S, Kavurt S, Balaban Adim S. Трансфузионно-ассоциированная реакция «трансплантат против хозяина» при тяжелом комбинированном иммунодефиците. J Investig Allergol Clin Immunol. 2010;20:153–156. [PubMed] [Академия Google]
62. Като М., Кимура Х., Секи М., Шимада А., Хаяши Ю., Морио Т. и соавт. Синдром Оменна — обзор нескольких фенотипов синдрома Оменна и мутаций RAG1/RAG2 в Японии. Аллергол Интерн. 2006; 55:115–9. [PubMed] [Google Scholar]
63. Kesserwan C, Sokolic R, Cowen EW, Garabedian E, Heselmeyer-Haddad K, Lee C-CR, et al. Мультицентровая выбухающая дерматофибросаркома у пациентов с тяжелым комбинированным иммунодефицитом с дефицитом аденозиндезаминазы. J Аллергия Клин Иммунол. 2012;129:762–9.e1. [Бесплатная статья PMC] [PubMed] [Google Scholar]
64. Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Клиническая картина синдрома Гришелли 2 типа и спектр мутаций RAB27A. Детский рак крови. 2010; 54: 563–72. [PubMed] [Google Scholar]
65. Kaplan J, De Domenico I, Ward DM. Синдром Чедиака-Хигаси. Карр Опин Гематол. 2008; 15:22–9. [PubMed] [Google Scholar]
Kaplan J, De Domenico I, Ward DM. Синдром Чедиака-Хигаси. Карр Опин Гематол. 2008; 15:22–9. [PubMed] [Google Scholar]
66. Dessinioti C, Stratigos AJ, Rigopoulos D, Katsambas AD. Обзор генетических нарушений гипопигментации: уроки биологии меланоцитов. Опыт Дерматол. 2009 г.;18:741–9. [PubMed] [Google Scholar]
67. Federici S, Sormani MP, Ozen S, Lachmann HJ, Amaryan G, Woo P, et al. Критерии предварительной клинической классификации аутовоспалительных периодических лихорадок, основанные на доказательствах. Энн Реум Дис. 2015; 74: 799–805. [PubMed] [Google Scholar]
68. Бэррон К.С., Кастнер Д.Л. Синдромы периодической лихорадки и другие наследственные аутовоспалительные заболевания. В: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR, редакторы. Учебник детской ревматологии. 7-е изд. Филадельфия, США: Эльзевир Сондерс; 2015. С. 609.–26. [Google Scholar]
69. Омбрелло А.К., Кастнер Д.Л. Наследственные синдромы периодической лихорадки и другие системные аутовоспалительные заболевания. В: Behrman RE, Kleigman RM, St. Geme JW III, Stanton BF, Schor NF, редакторы. Учебник Нельсона по педиатрии. 20-е изд. Филадельфия, США: Эльзевир Сондерс; 2015. С. 1193–1204. [Google Scholar]
В: Behrman RE, Kleigman RM, St. Geme JW III, Stanton BF, Schor NF, редакторы. Учебник Нельсона по педиатрии. 20-е изд. Филадельфия, США: Эльзевир Сондерс; 2015. С. 1193–1204. [Google Scholar]
70. Ким Х., Санчес Г.А.М., Гольдбах-Мански Р. Взгляд на менделевские интерферонопатии: сравнение CANDLE, SAVI с AGS, моногенной волчанкой. J Mol Med Berl Ger. 2016;94:1111–27. [Бесплатная статья PMC] [PubMed] [Google Scholar]
71. Crow YJ, Casanova J-L. STING-ассоциированная васкулопатия с началом в младенчестве — новая интерферонопатия. N Engl J Med. 2014; 371: 568–71. [PubMed] [Google Scholar]
72. Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Синдром Блау, прототип аутовоспалительной гранулематозной болезни. Pediatr Rheumatol Online J. 2014; 12:33. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Первичные иммунодефицитные заболевания | Адвокат Детская Больница
- Дом
- >
- Услуги
- >
- Первичное иммунодефицитное заболевание (PIDD)
- Услуги
Найдите врача
Если у ребенка есть инфекция или вирус, болезнь обычно проходит сама собой, и ребенок сразу же возвращается к действию. А вот детям с первичными иммунодефицитными заболеваниями (ПИ) прийти в норму не так-то просто. Наследственный сбой в их иммунной системе мешает детям с ПИ бороться с инфекциями. Многопрофильная команда педиатрических специалистов в детской больнице «Адвокат» имеет большой опыт ухода за детьми с ПИДД и стремится улучшить качество их жизни.
А вот детям с первичными иммунодефицитными заболеваниями (ПИ) прийти в норму не так-то просто. Наследственный сбой в их иммунной системе мешает детям с ПИ бороться с инфекциями. Многопрофильная команда педиатрических специалистов в детской больнице «Адвокат» имеет большой опыт ухода за детьми с ПИДД и стремится улучшить качество их жизни.
Иммунная система представляет собой сложную, точно сбалансированную систему, защищающую от инфекций. Если какая-либо часть этой системы изначально слаба или разбалансирована, ребенок становится восприимчивым к целому ряду серьезных, а иногда и смертельных инфекций и аутоиммунных проблем. Многие из них происходят от обычных организмов, которые у ребенка с полностью функционирующей иммунной системой обычно вызывают вялотекущую инфекцию или даже отсутствие заметных симптомов.
Дети с ПИ сталкиваются со своими особыми потребностями и трудностями. Наша преданная команда разрабатывает индивидуальные планы лечения для вашего ребенка, ежемесячно встречаясь, чтобы обсудить наилучший курс лечения. Хотя ПИ является хроническим заболеванием, с которым дети будут жить всю оставшуюся жизнь, наши специалисты стремятся помочь детям вырасти и жить полноценной независимой жизнью.
Хотя ПИ является хроническим заболеванием, с которым дети будут жить всю оставшуюся жизнь, наши специалисты стремятся помочь детям вырасти и жить полноценной независимой жизнью.
Что такое первичный иммунодефицит?
Первичный иммунодефицит (PI) — это наследственное заболевание, которое препятствует нормальному функционированию иммунной системы. Дети с ПИ рождаются без какой-либо иммунной защиты организма, что делает их более восприимчивыми к микробам, вызывающим инфекции. Дети, живущие с ПИ, часто страдают от инфекций, на которые не действуют лекарства, даже антибиотики.
Известно более 300 видов ИП. Они различаются по степени тяжести и симптомам для каждого человека. Помимо инфекций, дети с ПИ подвержены другим состояниям, таким как хронические заболевания легких и аутоиммунные заболевания, такие как артрит.
Признаки и симптомы PI
Признаки и симптомы варьируются от пациента к пациенту. Некоторые формы первичного иммунодефицита протекают очень легко и остаются незамеченными в течение многих лет. Другие формы более тяжелые и обнаруживаются при рождении. Наиболее частым признаком ПИДД у детей является повышенная уязвимость к инфекциям, которые не поддаются обычному лечению.
Другие формы более тяжелые и обнаруживаются при рождении. Наиболее частым признаком ПИДД у детей является повышенная уязвимость к инфекциям, которые не поддаются обычному лечению.
Некоторые признаки того, что у ребенка может быть ИП, включают:
- Рецидивирующие инфекции уха, легких, кожи или крови
- Аутоиммунные заболевания, поражающие суставы, кожу и желудочно-кишечный тракт
- Заболевания крови, необъяснимая анемия, низкий уровень лейкоцитов и низкий уровень тромбоцитов
- Хроническая диарея
- Задержка роста и развития
Почему стоит выбрать Детскую больницу Адвокат — Дубовая лужайка?
Детская больница Адвокат является признанным центром диагностики и лечения детей с ПИ. Наша программа лечения первичных иммунодефицитов является одной из двух в районе Чикаго, которые назначены центром PI Фонда Джеффри Моделла для диагностики и лечения.
Мы также обеспечиваем лидерство в мониторинге и изучении конкретных заболеваний PI через Фонд иммунодефицита. Почти все дети, которых мы лечим, внесены в реестр USIDNET. USIDNET (Сеть иммунодефицитов США) — это организация, созданная для продвижения научных исследований в области ИП. Мы являемся членом Общества клинической иммунологии и активно сотрудничаем с Национальным институтом здравоохранения в отношении некоторых редких заболеваний ПИ. Детская больница «Адвокат» также находится в авангарде поиска новой информации о первичных иммунодефицитных заболеваниях и методах лечения.
Почти все дети, которых мы лечим, внесены в реестр USIDNET. USIDNET (Сеть иммунодефицитов США) — это организация, созданная для продвижения научных исследований в области ИП. Мы являемся членом Общества клинической иммунологии и активно сотрудничаем с Национальным институтом здравоохранения в отношении некоторых редких заболеваний ПИ. Детская больница «Адвокат» также находится в авангарде поиска новой информации о первичных иммунодефицитных заболеваниях и методах лечения.
Наша команда в рамках междисциплинарной программы лечения синдрома Ди Джорджи лечит многих детей с синдромом Ди Джорджи. В команду входят врачи-кардиологи из всемирно известной детской кардиологической программы, педиатры по развитию, генетики, логопеды, физиотерапевты, детские психологи и диетологи.
Дети, направленные в Детскую больницу Адвоката с подтвержденным диагнозом ПИ, а также те, у кого есть подозрение на ПИ, проходят всестороннее обследование и лечение. Врачи в детской больнице Advocate понимают, что раннее выявление заболевания имеет решающее значение для здоровья каждого ребенка в долгосрочной перспективе, поэтому они повышают осведомленность местных педиатров, медсестер, жителей и других медицинских работников посредством обучения и обучения. Наши врачи обучены распознавать настораживающие признаки ПИ и наблюдать за ними при поступлении детей в больницу.
Наши врачи обучены распознавать настораживающие признаки ПИ и наблюдать за ними при поступлении детей в больницу.
Кто будет в команде по уходу за моим ребенком?
Детская больница Advocate имеет персонал с обширными знаниями и знаниями. Работая вместе и собираясь ежемесячно, чтобы определить лучший курс лечения для каждого ребенка, наша исключительная команда включает в себя:
- Детский иммунолог: Лечит различные заболевания и нарушения иммунной системы.
- Детский инфекционист: специализируется на лечении сложных инфекций. Специалист лечит широкий спектр инфекционных и иммунологических заболеваний, вызванных бактериями, вирусами, грибками и паразитами.
- Детский кардиолог: лечит легкие и сложные проблемы с сердцем у младенцев и детей.
- Детский гастроэнтеролог: лечит детей с проблемами пищеварения, печени и питания, такими как пищевая аллергия, воспалительные заболевания кишечника или панкреатит.
- Детский гематолог/онколог: специально обучен для лечения заболеваний крови.

- Детский пульмонолог: диагностирует и лечит состояния и заболевания легких, такие как астма.
- Детский психолог: изучает умственное, социальное и эмоциональное развитие ребенка с ПИ.
- Генетический консультант: работает с семьями, координируя тестирование, интерпретируя результаты и рассматривая все доступные варианты дополнительного тестирования, наблюдения, хирургического вмешательства или исследования.
- Зарегистрированный диетолог: эксперт по пищевым продуктам и питанию, который занимается лечением и профилактикой различных заболеваний, разрабатывая план диеты для вашего ребенка.
- Медсестра-координатор-исследователь: курирует клиническую помощь детям с ПИ.
- Социальный работник: поддерживает вашего ребенка и всю вашу семью, работая с вами, чтобы понять болезнь. Социальный работник помогает семье понять болезнь и консультирует по поводу решений, которые необходимо принять.
Какие варианты лечения доступны?
Лечение ПИ зависит от заболевания. Варианты могут включать нацеливание на определенные инфекции или различные формы терапии для укрепления иммунной системы.
Варианты могут включать нацеливание на определенные инфекции или различные формы терапии для укрепления иммунной системы.
Дети в детской больнице Адвокат имеют доступ ко всему спектру лечения ИПН, например:
- Антибиотики и другие лекарства для профилактики и лечения инфекций
- Иммуноглобулиновая терапия для укрепления иммунной системы
- Терапия гамма-интерфероном для улучшения функции нейтрофилов
- Терапия для повышения уровня иммуностимулирующих лейкоцитов
- Терапия моноклональными антителами, такая как Abatacept
- Оценка трансплантации стволовых клеток для лечения некоторых форм иммунодефицита
Профилактика инфекций
Хотя первичные иммунодефицитные заболевания являются генетическими и не могут быть предотвращены, родители могут принять меры, чтобы их дети с ослабленной иммунной системой не заразились инфекциями:
- Гигиена: регулярно мыть руки и чистить зубы два раза в день
- Питание: соблюдайте здоровую сбалансированную диету
- Воздействие: избегайте скопления людей и людей с простудой или другими инфекциями
- Лекарства: не забывайте регулярно принимать лекарства
- Вакцинация: узнайте, какие прививки должен получить ваш ребенок
Для детей.