Митохондриальная патология: Митохондриальная патология у детей | #01/16

Митохондриальные эпилепсии
10 Февраля 2019г.
Митохондриальные заболевания представлены большой группой. Они развиваются в результате нарушения нормального функционирования митохондрий – главного поставщика энергии в клетки. Эти нарушения могут быть избирательными и затрагивать различные системы организма. Если изменения происходят в тканях и органах нервной системы, то развивается митохондриальная эпилепсия.
Причины болезни
Митохондриальная эпилепсия является следствием дефекта митохондрий, который может быть обусловлен генетическими, биохимическими или структурными изменениями. Они могут затрагивать различные звенья, которые участвуют в передаче энергии, но результат всегда будет одинаковым – сниженная продукция АТФ – основного источника энергии для всех клеточных процессов. В результате развивается нестабильность потенциала на мембранах нервных клеток, из-за чего они погибают. Их место занимают новые клетки, но несмотря на дефицит энергии, они склонны к постоянной активности. Такие изменения приводят к появлению патологического очага возбуждения в головном мозге, который и является причиной развития митохондриальной эпилепсии.
Такие изменения приводят к появлению патологического очага возбуждения в головном мозге, который и является причиной развития митохондриальной эпилепсии.
Диагностика заболевания
Митохондриальные заболевания могут быть схожи с другими видами мультисистемной патологии, поэтому их необходимо отличать друг от друга. Точку в этом вопросе может поставить генетическое тестирование, которое поможет установить характерный фенотип или специфическую мутацию. Обычно назначают секвенирование ядерной или митохондриальной ДНК. Данный метод точен и информативен для анализа индивидуальной генетической изменчивости. С его помощью можно расшифровать генетический код, который уникален для каждого индивидуума. Выбор конкретного метода исследования зависит от клинических проявлений у пациента. При несиндромальной митохондриальной эпилепсии предпочтение отдается секвенированию ядерной ДНК, в остальных случаях может назначаться секвенирование митохондриальной ДНК. Таким образом, оба метода применяются при диагностике заболевания, и они доступны в медико-генетическом центре «Геномед».
Среди других методов диагностики могут применяться:
- общеклинические анализы. В крови пациентов с митохондриальной эпилепсией выявляется повышенный уровень молочной кислоты (лактата), ацилкарнитина, кетоновых тел, в моче – повышение концентрации органических кислот;
- биопсия мышц. Позволяет получить митохондрии и изучить активность ферментов дыхательной цепи;
- КТ или МРТ головного мозга. Применяется для исключения опухолевой природы эпилепсии. Иногда специалистам удается выявить очаговые изменения в головном мозге, которые могут быть следствием метаболической энцефалопатии;
- ЭЭГ. Позволяет оценить электрическую активность мозга, выявить очаги патологической импульсации.
Диагностика заболевания также основывается и на клинической картине. У пациентов с митохондриальной эпилепсией часто отмечаются психические расстройства, мышечная слабость, быстрая утомляемость, светобоязнь и др.
Лечение митохондриальной эпилепсии
Эффективной терапии, которая могла бы воздействовать на причину митохондриальной эпилепсии, пока не существует.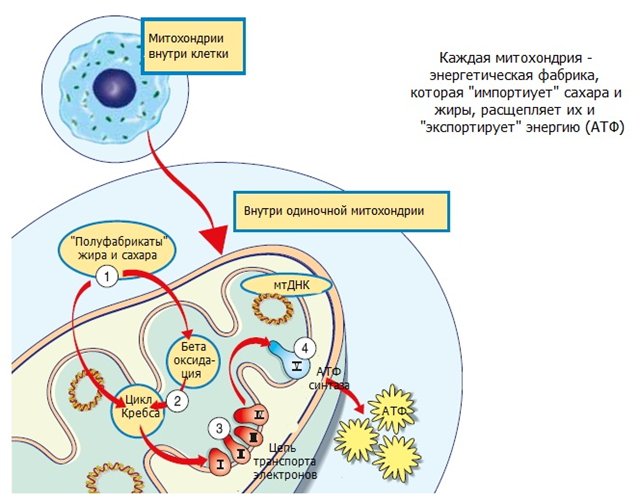 Пациенты получают симптоматическое лечение. Дополнительно назначаются препараты, которые снижают уровень лактата в крови, витамины группы В, аскорбиновая кислота, токоферол, янтарная кислота. Если митохондриальная эпилепсия сочетается с патологией других органов, то схема лечения дополняется необходимыми лекарственными препаратами.
Пациенты получают симптоматическое лечение. Дополнительно назначаются препараты, которые снижают уровень лактата в крови, витамины группы В, аскорбиновая кислота, токоферол, янтарная кислота. Если митохондриальная эпилепсия сочетается с патологией других органов, то схема лечения дополняется необходимыми лекарственными препаратами.
Прогноз во многом зависит от того, насколько своевременно была выявлена митохондриальная эпилепсия. В благоприятных случаях удается купировать основные симптомы и улучшить качество жизни пациентов, несмотря на необходимость пожизненного лечения.
Глаукома как митохондриальная патология | Фролов М.А.
M.A. Frolov
Russian University of Peoples’ Friendship, Medical Faculty, Department of Eye Diseases, Moscow
Author discusses mitochondrial role in pathogenesis of ophthalmologic pathologies, questions of their diagnostics and approaches to their treatment.
В настоящее время доказанным считается тот факт, что наиболее ранние повреждения при глаукоме происходят в митохондриях аксонов ганглиозных клеток сетчатки (ГКС). Установлена ведущая роль митохондрий в старении, апоптозе и нейродегенеративных расстройствах, к которым в последнее время относят и глаукому [2]. Предполагается, что целый ряд взаимодействующих факторов может вносить вклад в нейрональные повреждения при нейродегенеративных расстройствах, и одним из них является вторичная дисфункция митохондрий. Исходя из этого, прогрессирование глаукомы и появление глаукоматозной оптической нейропатии (ГОН), а затем и атрофии могут рассматриваться как митохондриальная патология [1,2,8]. Следовательно, для того чтобы разобраться в патогенезе ГОН, необходимо более детально рассмотреть митохондрии и происходящие в них процессы.
Установлена ведущая роль митохондрий в старении, апоптозе и нейродегенеративных расстройствах, к которым в последнее время относят и глаукому [2]. Предполагается, что целый ряд взаимодействующих факторов может вносить вклад в нейрональные повреждения при нейродегенеративных расстройствах, и одним из них является вторичная дисфункция митохондрий. Исходя из этого, прогрессирование глаукомы и появление глаукоматозной оптической нейропатии (ГОН), а затем и атрофии могут рассматриваться как митохондриальная патология [1,2,8]. Следовательно, для того чтобы разобраться в патогенезе ГОН, необходимо более детально рассмотреть митохондрии и происходящие в них процессы.
Митохондрии представляют собой мембранную органеллу клетки, основной функцией которой является выработка энергии в процессе дыхания и метаболизма в виде АТФ – универсальной энергетической единицы. Число митохондрий в клетке непостоянно и зависит от ее природы – чем выше энергетические потребности клетки, тем больше митохондрий в ней содержится. Совокупность всех митохондрий в клетке называется хондриом.
Совокупность всех митохондрий в клетке называется хондриом.
Длина этой органеллы колеблется от 1 до 70 мкм, ширина – 0,25–1,0 мкм, диаметр не превышает 1 мкм. По форме митохондрии могут быть спиральными, округлыми, вытянутыми, чашевидными, разветвленными. Митохондрии способны изменять форму, а также перемещаться в особо активные участки клетки, используя структуры цитоскелета. Такое перемещение позволяет клетке сосредоточить большее число митохондрий в зонах наибольшего энергопотребления. В других случаях положение митохондрий более постоянно.
Каждая митохондрия окружена оболочкой, состоящей из двух мембран. Наружную мембрану от внутренней отделяет межмембранное пространство. Внутренняя мембрана образует многочисленные гребневидные складки – так называемые кристы, которые существенно увеличивают поверхность внутренней мембраны, обеспечивая место для размещения компонентов дыхательной цепи. Через внутреннюю мембрану осуществляется активный транспорт АДФ и АТФ. Митохондрии содержат кольцевую ДНК и 70S-рибосомы [7]. В норме ДНК всех митохондрий в клетке имеет одинаковую структуру – это состояние называется гомоплазмия [3].
В норме ДНК всех митохондрий в клетке имеет одинаковую структуру – это состояние называется гомоплазмия [3].
В митохондриальном геноме могут происходить мутации, что приводит к появлению митохондрий с нарушенной функцией. При этом возникает гетероплазмия – в клетке одновременно присутствуют митохондрии с мутированной и немутированной ДНК [3,6]. Нормальная ДНК может компенсировать патологический эффект мутации, и клетка функционирует за счет неизмененных митохондрий до тех пор, пока получает достаточно энергии. В этот период клинических симптомов нарушения митохондриальных функций не отмечается. Таким образом, митохондриальные заболевания могут иметь длительное бессимптомное течение.
При снижении энергопродукции в клетке начинается компенсаторная пролиферация всех митохондрий, включая дефектные. Это влечет за собой накопление дефектных форм, количество которых впоследствии становится достаточным для проявления патологических признаков – происходит манифестация заболевания. Стоит отметить, что при раннем начале заболевание имеет более тяжелое течение и неблагоприятный прогноз.
Стоит отметить, что при раннем начале заболевание имеет более тяжелое течение и неблагоприятный прогноз.
Клинические проявления патологий, в основе которых лежат митохондриальные мутации, достаточно разнообразны. Несмотря на то, что чаще поражаются органы с высокой метаболической активностью, такие как нейроны головного мозга, скелетная мускулатура, миокард, клетки костного мозга и эндокринные железы, хронический недостаток энергии может привести к изменениям в любом органе [1].
Особый интерес представляет предположение о возможном общем механизме развития патологических изменений в структурах головного мозга и органа зрения. Учитывая, что сетчатка глаза, зрительные пути и центральная нервная система (ЦНС) являются единым целым, можно предположить наличие митохондриальной патологии, усугубляющей течение и развитие не только заболеваний ЦНС, но и непосредственно глазного яблока, особенно имеющих хронический неизлечимый характер [1]. В связи с этим становится актуальным изучение состояния митохондрий клеток различных структур глаза, таких как трабекула, Шлеммов канал, сетчатка, зрительный нерв, а также более высоких отделов зрительного пути – вплоть до структур коры головного мозга [2–4].
Среди заболеваний органа зрения с доказанной митохондриальной патологией особо выделяется атрофия зрительного нерва Лебера. Данное заболевание начинается, как правило, в возрасте от 18 до 30 лет, и проявляется быстро или постепенно развивающимся двусторонним снижением центрального зрения [3,6]. Атрофия зрительного нерва также появляется и при других заболеваниях зрительного нерва мультифокальной природы, таких как оптический неврит, передняя ишемическая оптическая нейропатия, ГОН.
На сегодняшний день описаны десятки вариантов мутаций митохондриального генома, приводящие к развитию атрофии зрительного нерва. Так, было проведено предварительное изучение состояния митохондрий аксонов зрительного нерва при экспериментальной адреналин-индуцированной глаукоме у кроликов. В результате исследования волокон зрительного нерва у экспериментальных животных были обнаружены изменения митохондрий различной степени выраженности у 97% органелл. Митохондрии были резко увеличены в размерах, имелись просветления матрикса и вакуоли, кристы были укорочены, редуцированы и расположены вблизи мембран. В единичных митохондриях также отмечались явления дегенерации и деструкции [2].
В единичных митохондриях также отмечались явления дегенерации и деструкции [2].
Вследствие нарушения структуры митохондрий происходит также и нарушение ее функционирования: изменяется проницаемость мембран и повышается приток ионов кальция, что отражается на мембранном потенциале; становятся более интенсивными процессы окисления, образуется большое количество АФК (активных форм кислорода, токсичных для клетки) и оксида азота. Данные нарушения приводят к запуску механизма апоптоза – добровольной самоликвидации клетки с целью остановки неправильной разрушительной работы митохондрии и предотвращения риска мутации ДНК. Именно апоптоз на данный момент считается ключевым моментом в патогенезе глаукоматозной атрофии при первичной открытоугольной глаукоме [4,5].
Новое понимание процессов развития глаукоматозного процесса дает возможность искать новые средства диагностики данной патологии и прогноза ее развития. Так, важной становится оценка функций митохондрий. Для этого в настоящее время используют клинические, морфологические (гистохимическое изучение активности ферментов митохондрии и распределение в ней различных субстратов – липидов, гликогена, солей кальция – в биоптатах тканей), биохимические (оценка уровней лактата и пирувата, антиоксидантной активности, продуктов перекисного окисления липидов крови) и молекулярно-генетические методы (выявление мутаций митохондриальной ДНК и ядерных мутаций, приводящих к нарушению синтеза митохондриальных белков). Кроме того, молекулярно-генетические методы позволяют дифференцировать оптические нейропатии различного генеза [3,6,8].
Кроме того, молекулярно-генетические методы позволяют дифференцировать оптические нейропатии различного генеза [3,6,8].
Кроме диагностики встает вопрос о коррекции медикаментозного лечения патологий с митохондриальным компонентом в патогенезе. На данный момент применяется только симптоматическая терапия, основными направлениями которой являются повышение эффективности энергообмена в тканях и предупреждение повреждения митохондриальных мембран. Для этого применяются витамины (тиамин, рибофлавин, никотинамид, коэнзим Q10, витамин С, цитохром С), антиоксиданты (витамин Е, альфа-липоевая кислота) и мембранопротекторы (метионин, цитиколин, убихинон, эссенциальные фосфолипиды и др.). Лечение включает также альтернативные источники энергии (креатин моногидрат), стратегию снижения уровня лактата (дихлорацетат) и физические упражнения [1–3].
Литература
1. Алексеев В.Н., Мартынова Е.Б. и др. Значение митохондриальной патологии в медицине и в офтальмологии (обзор) // Российская глаукомная школа. Глаукома: теория и практика: Сб. научн. труд. – СПб., 2011. – С. 5–11.
Глаукома: теория и практика: Сб. научн. труд. – СПб., 2011. – С. 5–11.
2. Алексеев В.Н., Газизова И.Р., Никитин Д.Н. Роль нарушений энергетического обмена в прогрессировании первичной открытоугольной глаукомы // РООФ: Сб. научн. труд. – М., 2011. – С. 210–213.
3. Вельтищев Ю.Е., Темин П.А. Митохондриальные болезни. Наследственные болезни нервной системы // Медицина. – 1998. – № 4. – С. 346–409.
4. Курышева Н.И. Глаукоматозная оптическая нейропатия. – М.: Медпресс-информ, 2006. – С. 13–29.
5. Green D.R., Reed J.C. Mitochondria and apoptosis // Science. – 1998. – Vol. 281. – Р. 1309–1316.
6. Schon T.A., Bonilla T., DiMauro S. Mitochondrial DNA mutations and pathogenesis // J. Bioenergy. Biomembr. – 1997. – Vol. 29. – Р. 131–149.
7. Taylor D.J., Green N.P.O., Stout G.W. Biological science // Cambrige University Press. – 2002. – Р. 355–359.
8. Wallece D.C., Broun M.D., Lott M.L. Mitochondrial DNA variation in human evolution and deasease // Gene. – 1999. – Vol. 238. – Р. 211–230.
– 1999. – Vol. 238. – Р. 211–230.
митохондриальная болезнь | патология | Британника
- Похожие темы:
- генетическое заболевание человека
метаболическое заболевание
митохондрия
митохондриальное расстройство дыхательной цепи
См. все связанные материалы →
митохондриальное заболевание , также называемое митохондриальным заболеванием , любое из нескольких сотен наследственных заболеваний, возникающих в результате функциональной недостаточности митохондрий, типа клеточных органелл. Митохондриальные заболевания могут возникнуть в любом возрасте и чрезвычайно разнообразны по своим клиническим и молекулярным особенностям. Они варьируются по степени тяжести от относительно легкого заболевания, поражающего только один орган, до изнурительного, а иногда и смертельного заболевания, поражающего несколько органов. Широкий спектр симптомов создает серьезные проблемы для диагностики состояний, связанных с митохондриальной дисфункцией. По крайней мере, 1 из каждых 5000 человек во всем мире страдает митохондриальным заболеванием.
По крайней мере, 1 из каждых 5000 человек во всем мире страдает митохондриальным заболеванием.
Хотя некоторые митохондриальные заболевания вызываются мутациями в митохондриальном геноме (мтДНК), большинство состояний являются результатом мутаций в генах ядерного генома, который кодирует ряд белков, которые экспортируются и транспортируются в митохондрии в клетка. Белки собираются в митохондриях, образуя цепь переноса электронов (ЭТЦ), первичный аппарат генерации энергии в клетках. Перенос электронов от одного белкового компонента к другому в конечном итоге позволяет клеткам производить энергию в форме аденозинтрифосфата (АТФ), которая является основной формой энергии, используемой клетками и органами в организме. Дефицит любого из белков, входящих в состав ЭТЦ, может нарушить выработку АТФ и привести к накоплению неиспользуемых промежуточных продуктов (начальных молекул сахара и жира, которые входят в ЭТЦ) и активных форм кислорода (АФК; свободный радикал, содержащий кислород). Неиспользованные промежуточные соединения могут реагировать с другими молекулами, что приводит к образованию вредных побочных продуктов, таких как молочная кислота, в то время как АФК могут реагировать с различными клеточными молекулами, вызывая окислительный стресс и гибель клеток.
Неиспользованные промежуточные соединения могут реагировать с другими молекулами, что приводит к образованию вредных побочных продуктов, таких как молочная кислота, в то время как АФК могут реагировать с различными клеточными молекулами, вызывая окислительный стресс и гибель клеток.
Признаки и симптомы митохондриального заболевания различаются в зависимости от пораженного органа или систем органов. Возможные показания включают задержку развития, снижение роста, утомляемость, мигрень, мышечную слабость, мышечную боль, кардиомиопатию, печеночную недостаточность, слепоту, атрофию зрительного нерва (дегенерацию зрительного нерва), потерю слуха, диабет и судороги. Часто появляются группы признаков и симптомов, указывающие на дискретный синдром. Например, у лиц в возрасте от трех месяцев до двух лет для синдрома Ли характерны задержка физического развития, прогрессирующая неврологическая дегенерация (со снижением мышечного тонуса, некоординированными движениями и непроизвольными и повторяющимися сокращениями мышц), а также проблемы со зрением, дыханием и сердцем. . Синдром Кернса-Сейра, с другой стороны, характеризуется в первую очередь прогрессирующей слабостью или параличом глазных мышц и ретинопатией (повреждением светочувствительной сетчатки глаза), что может привести к опущению век и потере зрения; начало обычно в возрасте до 20 лет.
. Синдром Кернса-Сейра, с другой стороны, характеризуется в первую очередь прогрессирующей слабостью или параличом глазных мышц и ретинопатией (повреждением светочувствительной сетчатки глаза), что может привести к опущению век и потере зрения; начало обычно в возрасте до 20 лет.
Диагноз митохондриального заболевания основывается на клинических признаках и, по возможности, на результатах генетического тестирования. Семейный анамнез материнской семьи может дать важную диагностическую информацию, поскольку наследственные митохондриальные заболевания передаются от матери к ее потомству и передаются строго по материнской семейной линии. Лица, страдающие митохондриальными заболеваниями, могут получить генетическое консультирование для оценки риска передачи наследственного заболевания.
Лечение митохондриальных заболеваний является поддерживающим. Оптические приспособления, включая замену хрусталика, и слуховые аппараты, такие как кохлеарные имплантаты, могут помочь людям с нарушениями зрения или слуха. Некоторым пациентам могут помочь имплантированные кардиостимуляторы или дефибрилляторы. Поддерживающая терапия дефицита определенных компонентов ETC может включать пероральное введение таких веществ, как коэнзим Q 10 , L-креатин (моногидрат креатина) или рибофлавин. Физические упражнения также могут помочь облегчить симптомы у некоторых людей.
Некоторым пациентам могут помочь имплантированные кардиостимуляторы или дефибрилляторы. Поддерживающая терапия дефицита определенных компонентов ETC может включать пероральное введение таких веществ, как коэнзим Q 10 , L-креатин (моногидрат креатина) или рибофлавин. Физические упражнения также могут помочь облегчить симптомы у некоторых людей.
Кара Роджерс
Митохондриальная патология — Нейродегенерация — Книжная полка NCBI
Ключевые моменты, поднятые отдельными докладчиками
Из-за высокой потребности в энергии нейроны особенно уязвимы к травмам и гибели из-за дисфункциональных митохондрий.
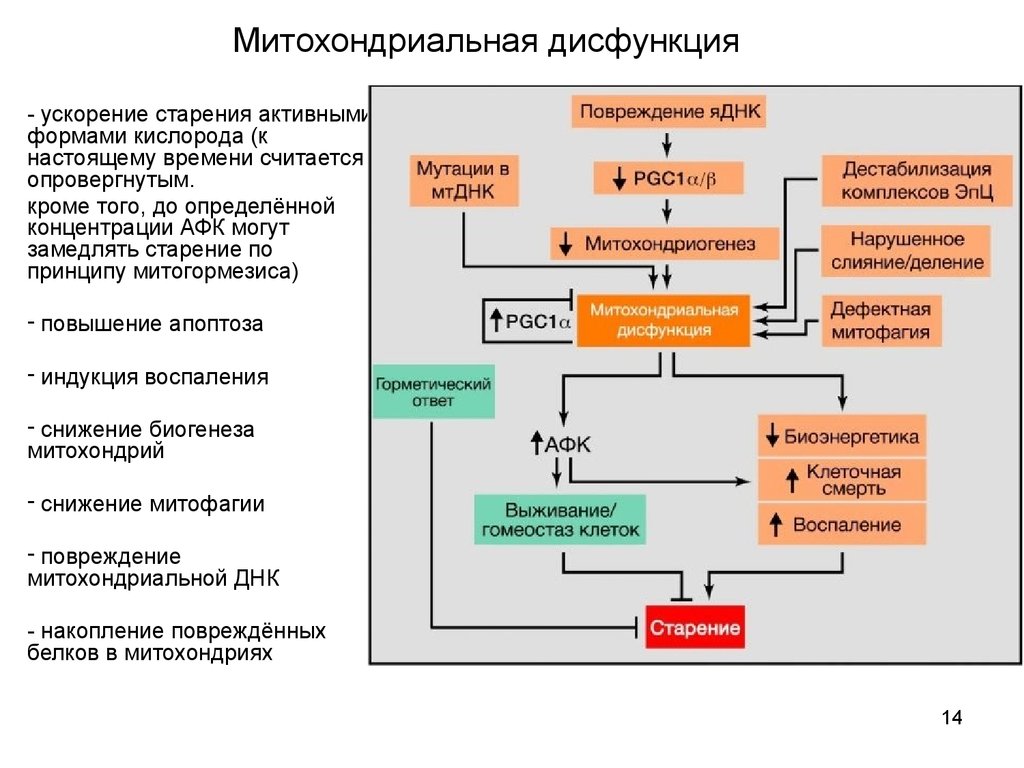
Патологические и физиологические данные свидетельствуют о дисфункции митохондрий при всех основных нейродегенеративных заболеваниях.
Остаются вопросы относительно того, является ли митохондриальная дисфункция причиной нейродегенеративного заболевания. Даже если это не является причиной, митохондриальная дисфункция все еще очень важна и, вероятно, способствует заболеванию.
 Идентификация методов лечения для улучшения митохондриальной функции или деградации дисфункциональных митохондрий может иметь смысл.
Идентификация методов лечения для улучшения митохондриальной функции или деградации дисфункциональных митохондрий может иметь смысл.Изучение первичных митохондриальных заболеваний может пролить свет на нейродегенеративные заболевания, которые проявляют сходную патологию. Поскольку оба типа заболеваний поражают множество путей и систем органов, они требуют подхода системной биологии.
Потенциальные терапевтические подходы включают лекарства, индуцирующие митохондриальный генез, каталитические антиоксиданты для защиты от активных форм кислорода, регуляторы внутриклеточного кальция и регуляторы окислительно-восстановительного потенциала через митохондриальную мембрану. Поддержание окислительно-восстановительного потенциала имеет решающее значение для целостности митохондрий и контроля окислительного фосфорилирования.
Митохондрии — это клеточные органеллы, ответственные за окислительное фосфорилирование, жизненно важный процесс превращения питательных веществ в молекулы аденозинтрифосфата (АТФ), которые обеспечивают энергию для нормального функционирования клеток. Каждый нейрон имеет по меньшей мере сотни митохондрий. Поскольку нервные клетки являются постмитотическими, любое стойкое митохондриальное повреждение будет накапливаться с возрастом и приводить к дисфункции. Широко распространенное повреждение митохондрий приводит к гибели клеток, потому что они больше не могут производить достаточно энергии. Действительно, сами митохондрии высвобождают ферменты, ответственные за гибель клеток. Мозг особенно уязвим для митохондриальной дисфункции, поскольку его энергетические потребности выше, чем у любого другого органа тела. Мозг составляет всего 2 процента массы тела, но при этом потребляет 20 процентов кислорода.
Каждый нейрон имеет по меньшей мере сотни митохондрий. Поскольку нервные клетки являются постмитотическими, любое стойкое митохондриальное повреждение будет накапливаться с возрастом и приводить к дисфункции. Широко распространенное повреждение митохондрий приводит к гибели клеток, потому что они больше не могут производить достаточно энергии. Действительно, сами митохондрии высвобождают ферменты, ответственные за гибель клеток. Мозг особенно уязвим для митохондриальной дисфункции, поскольку его энергетические потребности выше, чем у любого другого органа тела. Мозг составляет всего 2 процента массы тела, но при этом потребляет 20 процентов кислорода.
Функционирование митохондрий определяется двумя отдельными геномами, один в митохондриях, известный как митохондриальная ДНК (мтДНК), а другой в ядре. Митохондриальный геном кодирует 13 белков, каждый из которых жизненно важен для окислительного фосфорилирования. Ядерный геном кодирует около 1500 генов, участвующих в митохондриальной биологии, включая белки, необходимые для репликации мтДНК, транскрипции, трансляции и посттрансляционных модификаций.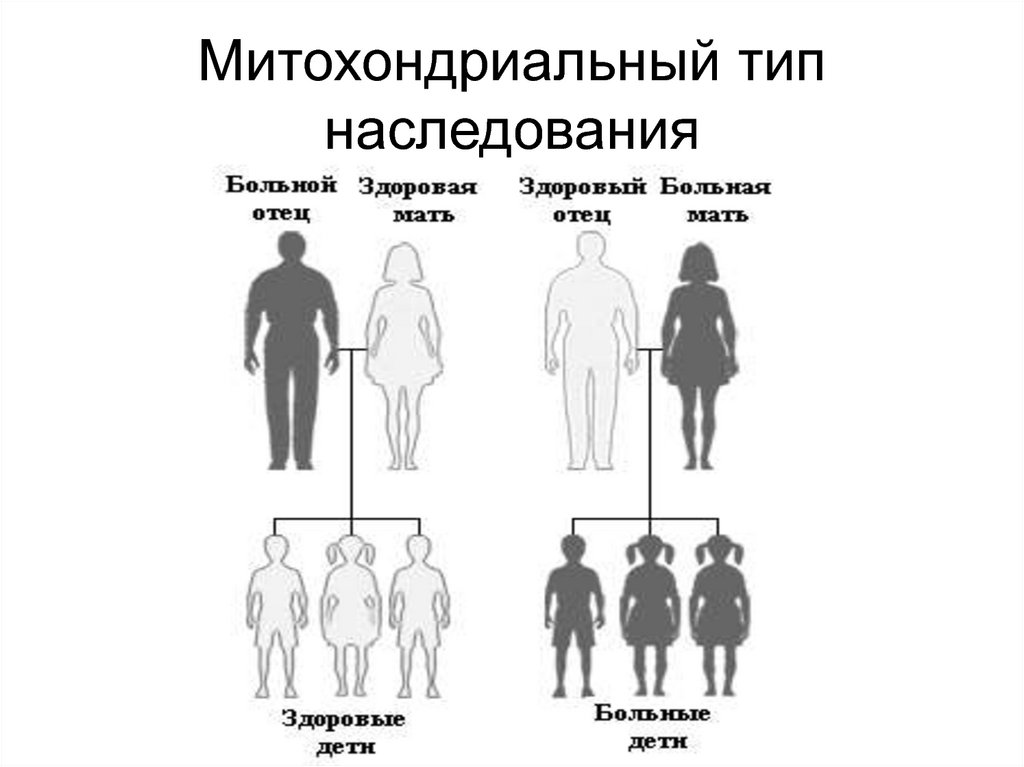 Существует только одна копия мтДНК, унаследованная от матери, по сравнению с двумя копиями ядерной ДНК, одна от матери, а другая от отца. Митохондрии не только ответственны за окислительное фосфорилирование, но также играют важную роль в метаболизме и передаче сигналов, включая синтез жирных кислот, метаболизм кетоновых тел, гомеостаз кальция и апоптоз. В частности, митохондрии обеспечивают большую часть клеточной энергии в форме АТФ. Они генерируют и регулируют активные формы кислорода, буферизуют уровни кальция внутри клетки и контролируют апоптоз (Wallace, 2005, 2010).
Существует только одна копия мтДНК, унаследованная от матери, по сравнению с двумя копиями ядерной ДНК, одна от матери, а другая от отца. Митохондрии не только ответственны за окислительное фосфорилирование, но также играют важную роль в метаболизме и передаче сигналов, включая синтез жирных кислот, метаболизм кетоновых тел, гомеостаз кальция и апоптоз. В частности, митохондрии обеспечивают большую часть клеточной энергии в форме АТФ. Они генерируют и регулируют активные формы кислорода, буферизуют уровни кальция внутри клетки и контролируют апоптоз (Wallace, 2005, 2010).
Митохондриальные дефекты обнаруживаются в патологических исследованиях всех основных нейродегенеративных заболеваний, сказал Вамси Мутха из Гарвардской медицинской школы. Диапазон митохондриальных дефектов включает фрагментацию и другие морфологические изменения, повышенную частоту мутаций в мтДНК, изменения проницаемости митохондриальных мембран, изменения окислительно-восстановительного потенциала, накопление мутантных белков и нарушение окислительного фосфорилирования (Reddy and Reddy, 2011).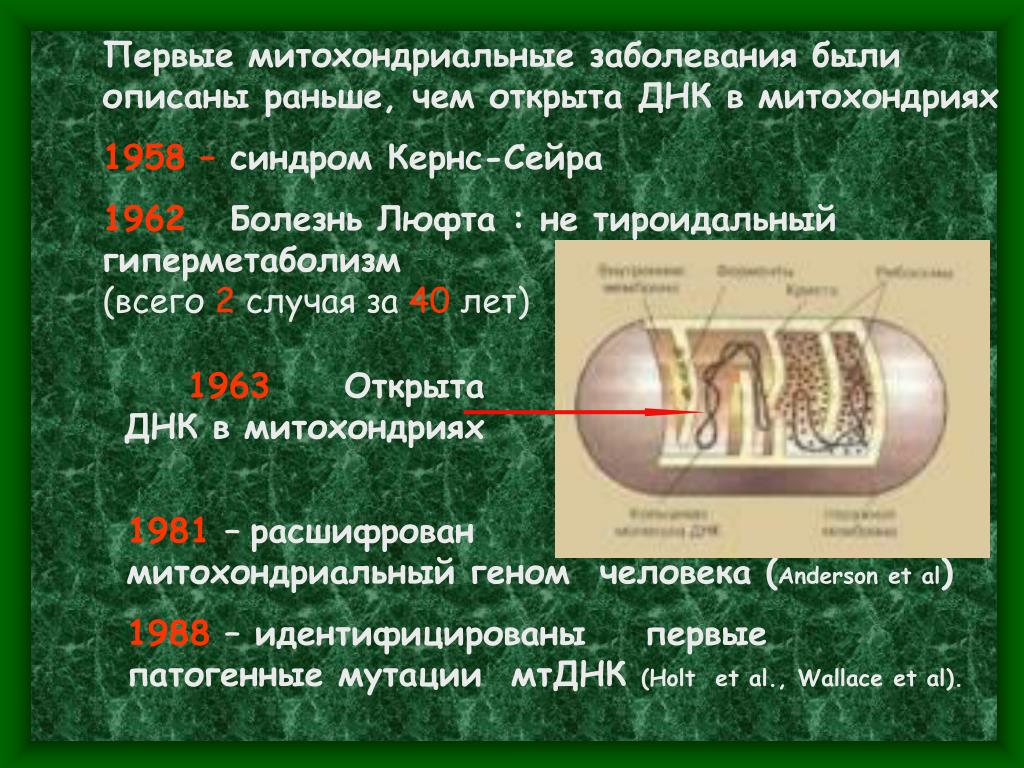 Но являются ли эти митохондриальные дефекты причиной нейродегенеративных заболеваний, является фундаментальным вопросом, сказал Мутха. Потенциальные роли митохондрий в нейродегенеративных заболеваниях, по его мнению, троякие: (1) они содержат первичные поражения и, таким образом, служат первичным источником патологии болезни; (2) они функционируют должным образом, но служат медиаторами или усилителями болезни; или (3) они являются сторонними наблюдателями, которые не способствуют патологии. Даже если митохондриальные дефекты не являются причиной, они, вероятно, способствуют этому, отмечает Нил Коуэлл из Бостонского университета, и поэтому любая терапия, которая сохраняет, улучшает или корректирует митохондриальную функцию, вероятно, будет полезна для предотвращения гибели клеток и прогрессирования заболевания.
Но являются ли эти митохондриальные дефекты причиной нейродегенеративных заболеваний, является фундаментальным вопросом, сказал Мутха. Потенциальные роли митохондрий в нейродегенеративных заболеваниях, по его мнению, троякие: (1) они содержат первичные поражения и, таким образом, служат первичным источником патологии болезни; (2) они функционируют должным образом, но служат медиаторами или усилителями болезни; или (3) они являются сторонними наблюдателями, которые не способствуют патологии. Даже если митохондриальные дефекты не являются причиной, они, вероятно, способствуют этому, отмечает Нил Коуэлл из Бостонского университета, и поэтому любая терапия, которая сохраняет, улучшает или корректирует митохондриальную функцию, вероятно, будет полезна для предотвращения гибели клеток и прогрессирования заболевания.
В этой главе обобщаются презентации семинаров, в которых приводятся доказательства митохондриальной дисфункции при основных нейродегенеративных заболеваниях. По словам нескольких участников, поскольку нет четких данных о том, является ли митохондриальная дисфункция причиной, может быть полезно взглянуть на первичные митохондриальные заболевания и применить системный подход к исследованиям.
МИТОХОНДРИАЛЬНАЯ ДИСФУНКЦИЯ И НЕЙРОДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ
Как отмечалось выше, митохондриальная дисфункция обнаруживается при основных нейродегенеративных заболеваниях. В этом разделе представлены презентации семинаров о митохондриальной дисфункции при болезни Паркинсона, боковом амиотрофическом склерозе (БАС), болезни Гентингтона и болезни Альцгеймера.
Болезнь Паркинсона
Болезнь Паркинсона характеризуется потерей дофаминсодержащих нейронов в области мозга, известной как черная субстанция. Патологические и другие исследования убедительно показали, что дефицит митохондрий накапливается в этой области мозга с возрастом, сказал Ричард Юл из Национального института неврологических расстройств и инсульта. Выступление Юла было сосредоточено на функции двух белков, которые мутируют при семейной болезни Паркинсона с ранним началом: Parkin и PINK1 (предполагаемая киназа 1, индуцированная PTEN).
Нормальные функции Parkin и PINK1 до недавнего времени не были хорошо изучены, сказал Юл. Накапливаются данные от нескольких видов о том, что эти белки обычно работают вместе, чтобы вызвать очищение поврежденных митохондрий, процесс, известный как митофагия. Само собой разумеется, что в случае мутации они могут не индуцировать митофагию, в результате чего дисфункциональные митохондрии накапливаются внутри клетки и вызывают гибель. Таким образом, нарушение митофагии связано с этиологией болезни Паркинсона с ранним началом (Narendra and Youle, 2011).
Накапливаются данные от нескольких видов о том, что эти белки обычно работают вместе, чтобы вызвать очищение поврежденных митохондрий, процесс, известный как митофагия. Само собой разумеется, что в случае мутации они могут не индуцировать митофагию, в результате чего дисфункциональные митохондрии накапливаются внутри клетки и вызывают гибель. Таким образом, нарушение митофагии связано с этиологией болезни Паркинсона с ранним началом (Narendra and Youle, 2011).
Когда митохондрии находятся в состоянии стресса или повреждены, заметил Юл, они накапливают PINK1. PINK1 представляет собой митохондриальный белок, обычно прикрепленный к внешней мембране митохондрии в низких концентрациях. Когда митохондриальная мембрана теряет свой электрический потенциал — будь то из-за мутаций ДНК, активных форм кислорода (АФК), 1 или других возмущений — PINK1 увеличивается. Увеличение концентрации PINK1, в свою очередь, способствует рекрутированию паркина, который представляет собой убиквитинлигазу, из цитозоля.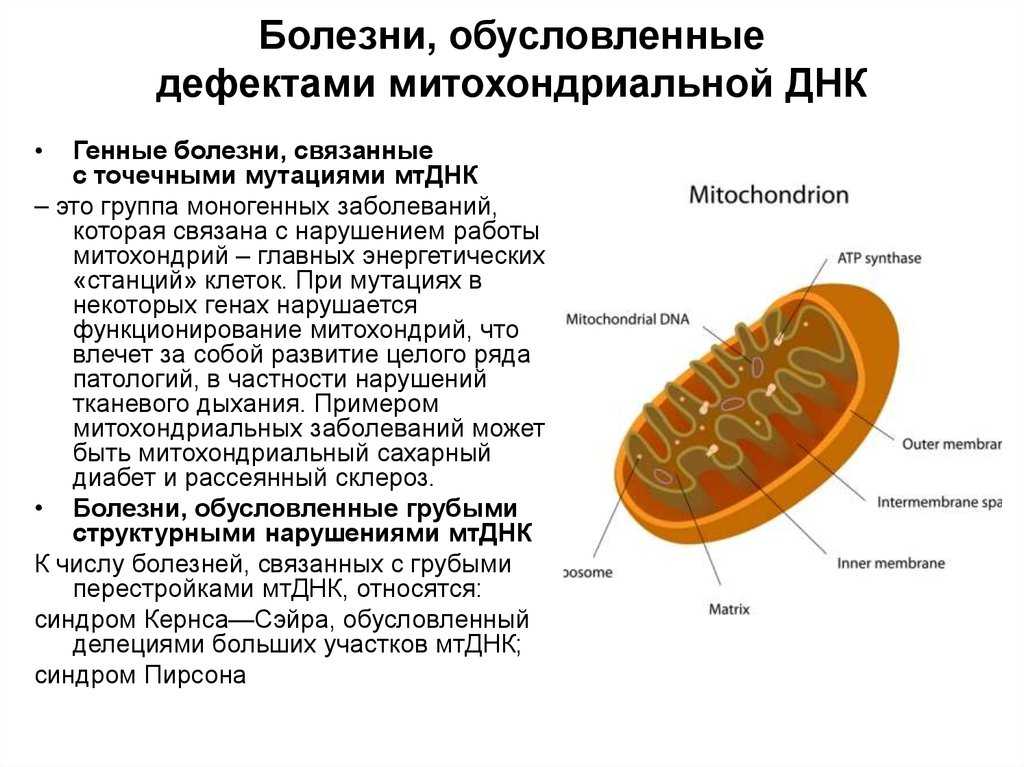 Паркин помечает поврежденную митохондрию убиквитином, процесс, запускающий образование аутофагосомы (см. также главу 3). Аутофагосома поглощает поврежденную митохондрию, затем сливается с лизосомой, которая разрушает ее. По словам Юла, результаты исследований PINK1/Parkin, которые были воспроизведены в нескольких лабораториях, открыли новый путь контроля качества митохондрий. Он отметил, что большая часть ранней работы в этой области была выполнена на культивируемых клеточных линиях, потому что соединения, используемые для индукции этого пути в культивируемых клеточных линиях, были слишком токсичны для нейронов. Однако совсем недавно две группы показали этот путь в нейронах (Cai et al., 2012; Wang et al., 2011).
Паркин помечает поврежденную митохондрию убиквитином, процесс, запускающий образование аутофагосомы (см. также главу 3). Аутофагосома поглощает поврежденную митохондрию, затем сливается с лизосомой, которая разрушает ее. По словам Юла, результаты исследований PINK1/Parkin, которые были воспроизведены в нескольких лабораториях, открыли новый путь контроля качества митохондрий. Он отметил, что большая часть ранней работы в этой области была выполнена на культивируемых клеточных линиях, потому что соединения, используемые для индукции этого пути в культивируемых клеточных линиях, были слишком токсичны для нейронов. Однако совсем недавно две группы показали этот путь в нейронах (Cai et al., 2012; Wang et al., 2011).
Юл сообщил, что его лаборатория начала программу скрининга наркотиков для выявления соединений, которые стимулируют путь PINK1/Parkin. Хотя он признал, что людям с болезнью Паркинсона с ранним началом может не помочь стимуляция пути, потому что их PINK1 или PARKIN мутированы, люди со спорадической болезнью Паркинсона могут получить пользу, как и другие люди с нейродегенеративным заболеванием, чьи митохондрии дисфункциональны.
Боковой амиотрофический склероз
БАС преимущественно поражает двигательные нейроны, приводя к прогрессирующей мышечной атрофии и параличу. В животных моделях БАС митохондриальные аномалии предшествуют симптомам болезни (Manfredi and Xu, 2005). Электронная микроскопия выявила структурные аномалии в митохондриях спинномозговых мотонейронов и в моторной коре у больных БАС. Нил Коуэлл из Бостонского университета сосредоточил свое выступление на SOD1 (супероксиддисмутаза Cu, Zn), первом идентифицированном гене, ответственном за возникновение БАС. Соответствующий белок мутирует примерно в 20% семейных случаев БАС. 2 Наиболее широко используемой животной моделью БАС является трансгенная мышь, несущая мутантный ген SOD1. У мыши развивается атрофия мышц, аналогичная таковой при БАС.
Митохондрии из двигательных нейронов в этой животной модели демонстрируют меньший размер, меньшее количество, дефект мембранного потенциала и нарушение слияния. Слияние митохондрий предназначено для распространения мтДНК среди популяции митохондрий и сохранения способности к окислительному фосфорилированию.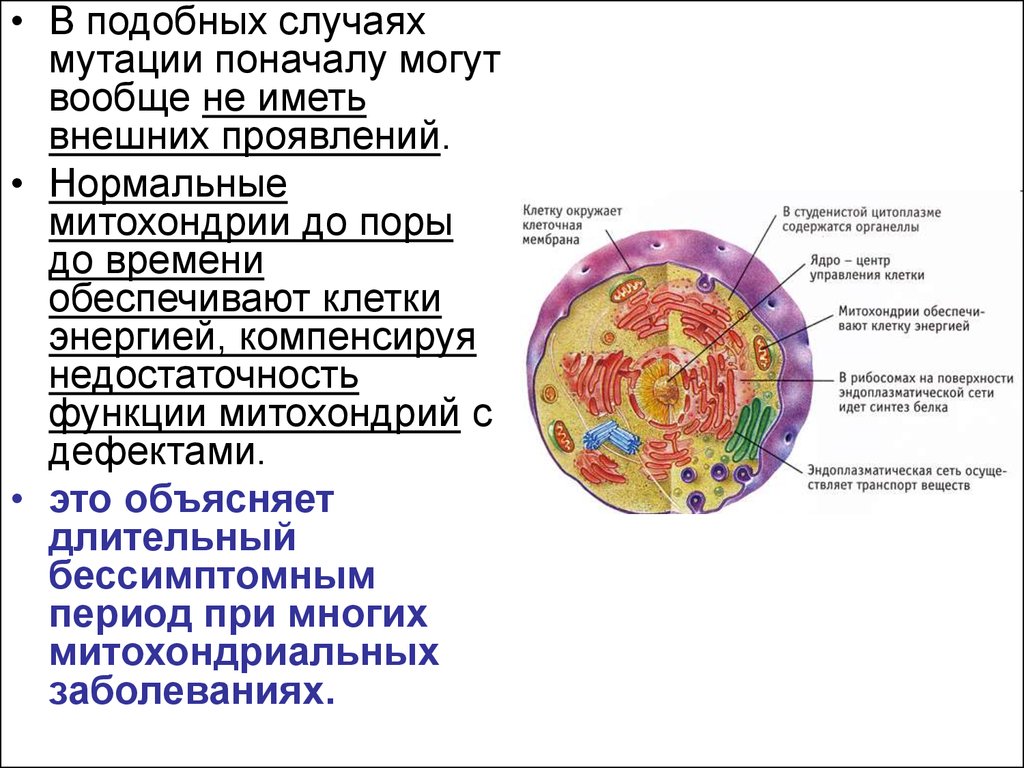 Эти морфологические и физиологические изменения в мотонейронах mutSOD1 не наблюдаются в мотонейронах SOD1 дикого типа (Magrane et al., 2012). mutSOD1 также изменяет уровни по крайней мере 50 различных митохондриальных белков, включая белки, участвующие в цепи переноса электронов и в их слиянии, указывая на возможный широко распространенный эффект mutSOD1 (Karbowski and Neutzner, 2012).
Эти морфологические и физиологические изменения в мотонейронах mutSOD1 не наблюдаются в мотонейронах SOD1 дикого типа (Magrane et al., 2012). mutSOD1 также изменяет уровни по крайней мере 50 различных митохондриальных белков, включая белки, участвующие в цепи переноса электронов и в их слиянии, указывая на возможный широко распространенный эффект mutSOD1 (Karbowski and Neutzner, 2012).
mutSOD1 также вызывает повреждение митохондрий, о чем свидетельствует увеличение содержания цитохрома с в цитозоле. Поскольку цитохром с является важным компонентом электрон-транспортной цепи, расположенной во внутренней мембране митохондрий, его выход в цитоплазму свидетельствует о разрушении мембран митохондрий. Но этот токсический эффект возникает только в присутствии белка Bcl-2, который может изменить свой функциональный фенотип и стать токсичным белком (Pedrini et al., 2010). Идентификация Bcl-2 как необходимого фактора токсичности SOD1 предполагает, что Bcl-2 можно использовать в качестве молекулярной мишени для препаратов, предназначенных для ингибирования его действия (Pedrini et al. , 2010). Bcl-2 также может быть важной мишенью не только при семейном БАС, но, возможно, и при спорадическом БАС, сказал Коуэлл. Это связано с тем, что исследования недавно показали, что в подгруппе пациентов с БАС с бульбарным началом SOD1 дикого типа становится гиперокисленной. Вместе с Bcl-2 гиперокисленный wtSOD1 проявляет митохондриальную токсичность, подобную наблюдаемой с mutSOD1. Таким образом, Bcl-2 представляет собой обычное связующее звено между семейным и подтипом спорадического БАС и, таким образом, является хорошей мишенью для терапевтических средств, которые его ингибируют.
, 2010). Bcl-2 также может быть важной мишенью не только при семейном БАС, но, возможно, и при спорадическом БАС, сказал Коуэлл. Это связано с тем, что исследования недавно показали, что в подгруппе пациентов с БАС с бульбарным началом SOD1 дикого типа становится гиперокисленной. Вместе с Bcl-2 гиперокисленный wtSOD1 проявляет митохондриальную токсичность, подобную наблюдаемой с mutSOD1. Таким образом, Bcl-2 представляет собой обычное связующее звено между семейным и подтипом спорадического БАС и, таким образом, является хорошей мишенью для терапевтических средств, которые его ингибируют.
Болезнь Гентингтона
Болезнь Гентингтона — это аутосомно-доминантное заболевание, при котором мутантный белок мхантингтин (mHTT) проявляет избыток полиглутаминовых повторов. mHTT локализуется на внешней митохондриальной мембране, где он оказывает обширное и вредное воздействие на митохондрии и избирательную потерю нейронов в стриатуме. Коуэлл сказал, что большое количество доказательств показывает, что mHTT снижает подвижность митохондрий, изменяет митохондриальную морфологию, вызывает дисрегуляцию кальция, снижает окислительное фосфорилирование и деполяризует митохондриальную мембрану в лимфобластах пациентов с болезнью Гентингтона. Деполяризация увеличивается с увеличением числа полиглутаминовых повторов. mHTT также изменяет баланс между слиянием и делением митохондрий (Lin and Beal, 2006; Reddy and Reddy, 2011).
Деполяризация увеличивается с увеличением числа полиглутаминовых повторов. mHTT также изменяет баланс между слиянием и делением митохондрий (Lin and Beal, 2006; Reddy and Reddy, 2011).
В последнее время появилось несколько терапевтических стратегий для лечения болезни Гентингтона, отметил Коуэлл. Одним из путей является нацеливание на белок митохондриального деления 3 , с которым связывается mHTT, белок 1, родственный GTPase dynamin (DRP1). Нацеленность на DRP1 подтверждается обнаружением того, что доминантно-негативный мутант DRP1K38A, который снижает активность DRP1, спасает митохондрии от следующих неблагоприятных эффектов mHTT: фрагментация митохондрий, дефекты антероградного и ретроградного транспорта митохондрий и гибель нейронов. Об этих результатах сообщалось в клетках людей с болезнью Хантингтона и мышей (Song et al., 2011). Другими словами, соединения, которые ингибируют DRP1, могут быть полезны в качестве потенциальной терапии.
Коуэлл описал еще два новых метода лечения. Первая направлена на детоксикацию HTT. Он включает внутрижелудочковую инфузию ганглиозида GM1, который фосфорилирует мутантный HTT по определенным аминокислотным остаткам серина. Такой подход не только снижал токсичность HTT, но и восстанавливал нормальную двигательную функцию у мышей с симптомами болезни Гентингтона (Di Pardo et al., 2012). Вторая терапия проводится уже одобренным препаратом меклизином. Этот препарат подавляет митохондриальное дыхание и активирует пути выживания клеток. В нескольких моделях болезни Хантингтона было обнаружено, что меклизин оказывает нейропротекторное действие (Gohil et al., 2011).
Первая направлена на детоксикацию HTT. Он включает внутрижелудочковую инфузию ганглиозида GM1, который фосфорилирует мутантный HTT по определенным аминокислотным остаткам серина. Такой подход не только снижал токсичность HTT, но и восстанавливал нормальную двигательную функцию у мышей с симптомами болезни Гентингтона (Di Pardo et al., 2012). Вторая терапия проводится уже одобренным препаратом меклизином. Этот препарат подавляет митохондриальное дыхание и активирует пути выживания клеток. В нескольких моделях болезни Хантингтона было обнаружено, что меклизин оказывает нейропротекторное действие (Gohil et al., 2011).
Болезнь Альцгеймера
Митохондриальная дисфункция предшествует патологическим изменениям, которые являются отличительными признаками болезни Альцгеймера (Yao et al., 2009). Дуглас Уоллес из Детской больницы Филадельфии предположил, что причиной болезни Альцгеймера и деменции в более широком смысле является основная дисфункция митохондрий. Начиная с 1993 года группа Уоллеса обнаружила мутацию в одном из генов митохондриальной тРНК. Мутация коррелировала с 3 процентами случаев болезни Альцгеймера с поздним началом, 5 процентами болезни Паркинсона и 7 процентами населения в целом. Вывод был позже подтвержден другими. Он утверждал, что тонкие дефекты в тРНК будут генерировать более глобальные дефекты синтеза митохондриальных белков. Впоследствии его группа начала изучать мутацию в положении 414 нуклеотида, которое примыкает к промотору контрольной области мтДНК. Ранее было показано, что мутация увеличивается с возрастом в фибробластах человека (Michikawa et al., 19).99). Команда Уоллеса обнаружила мутацию в 65% мозга больных Альцгеймером и в 57% мозга людей с синдромом Дауна и деменцией по сравнению с 0% контрольной группы того же возраста (Coskun et al., 2004).
Мутация коррелировала с 3 процентами случаев болезни Альцгеймера с поздним началом, 5 процентами болезни Паркинсона и 7 процентами населения в целом. Вывод был позже подтвержден другими. Он утверждал, что тонкие дефекты в тРНК будут генерировать более глобальные дефекты синтеза митохондриальных белков. Впоследствии его группа начала изучать мутацию в положении 414 нуклеотида, которое примыкает к промотору контрольной области мтДНК. Ранее было показано, что мутация увеличивается с возрастом в фибробластах человека (Michikawa et al., 19).99). Команда Уоллеса обнаружила мутацию в 65% мозга больных Альцгеймером и в 57% мозга людей с синдромом Дауна и деменцией по сравнению с 0% контрольной группы того же возраста (Coskun et al., 2004).
Совсем недавно команда Уоллеса исследовала более глобально контрольную область мтДНК в ткани, взятой из лобной коры головного мозга. Контрольная область отвечает за регуляцию транскрипции митохондриальных генов и помогает копировать мтДНК. Они обнаружили самый высокий уровень соматических мутаций в мозге людей с болезнью Альцгеймера (Coskun et al. , 2010). Частота мутаций также была повышена в случаях синдрома Дауна и деменции по сравнению с контрольной группой, но была ниже, чем при болезни Альцгеймера. В контрольной ткани действительно наблюдалось возрастное увеличение частоты мутаций, хотя уровень был ниже, чем в других группах. Повышенный уровень мутаций был также обнаружен в сыворотке и других тканях при болезни Альцгеймера и синдроме Дауна, что позволяет предположить, что это явление является системным. Но, по словам Уоллеса, мозг является наиболее пораженной тканью из-за его непропорционально высоких энергетических потребностей. Исследование также обнаружило снижение транскрипции мтДНК и уменьшение количества копий мтДНК, что указывает на снижение окислительного фосфорилирования.
, 2010). Частота мутаций также была повышена в случаях синдрома Дауна и деменции по сравнению с контрольной группой, но была ниже, чем при болезни Альцгеймера. В контрольной ткани действительно наблюдалось возрастное увеличение частоты мутаций, хотя уровень был ниже, чем в других группах. Повышенный уровень мутаций был также обнаружен в сыворотке и других тканях при болезни Альцгеймера и синдроме Дауна, что позволяет предположить, что это явление является системным. Но, по словам Уоллеса, мозг является наиболее пораженной тканью из-за его непропорционально высоких энергетических потребностей. Исследование также обнаружило снижение транскрипции мтДНК и уменьшение количества копий мтДНК, что указывает на снижение окислительного фосфорилирования.
Обращаясь к причинно-следственной связи нейродегенеративных заболеваний, Уоллес выразил мнение, что образование бляшек Aβ не является причинной; скорее, предположил он, белок Aβ изначально вырабатывается клетками в качестве компенсаторного средства защиты митохондрий. Но поскольку белок продолжает вырабатываться, он начинает агрегировать с образованием олигомеров и более крупных агрегатов, которые ингибируют митохондрии, что приводит к повреждению и гибели клеток. Согласно этой модели, белковые агрегаты способствуют гибели нейронов при нейродегенеративных заболеваниях, но не являются причиной. Первичная причина, согласно его гипотезе, лежит в дисфункциональных митохондриях. Он высказал мнение, что «общим патофизиологическим механизмом всех этих нейродегенеративных заболеваний является биоэнергетика». Затем в ходе дискуссии его допросили несколько скептически настроенных участников, которые не согласились с его причинной атрибуцией. В ответ Уоллес рассказал, как его команда разработала способ введения точечной мутации цитохромоксидазы в мтДНК и обнаружила, что у животного развилась кардиомиопатия, миопатия и патологические изменения в нейронах гиппокампа, ганглиозных клетках сетчатки и зрительном нерве. «Эта конкретная точечная мутация — она не имеет ничего общего с ядром — показывает, что энергетика может влиять на все эти различные функции», — заявил он.
Но поскольку белок продолжает вырабатываться, он начинает агрегировать с образованием олигомеров и более крупных агрегатов, которые ингибируют митохондрии, что приводит к повреждению и гибели клеток. Согласно этой модели, белковые агрегаты способствуют гибели нейронов при нейродегенеративных заболеваниях, но не являются причиной. Первичная причина, согласно его гипотезе, лежит в дисфункциональных митохондриях. Он высказал мнение, что «общим патофизиологическим механизмом всех этих нейродегенеративных заболеваний является биоэнергетика». Затем в ходе дискуссии его допросили несколько скептически настроенных участников, которые не согласились с его причинной атрибуцией. В ответ Уоллес рассказал, как его команда разработала способ введения точечной мутации цитохромоксидазы в мтДНК и обнаружила, что у животного развилась кардиомиопатия, миопатия и патологические изменения в нейронах гиппокампа, ганглиозных клетках сетчатки и зрительном нерве. «Эта конкретная точечная мутация — она не имеет ничего общего с ядром — показывает, что энергетика может влиять на все эти различные функции», — заявил он.
БЕЛКОВЫЕ ОТЛОЖЕНИЯ И ТОКСИЧНОСТЬ ДЛЯ МИТОХОНДРИЙ
Многочисленные данные свидетельствуют о том, что токсичные белки, такие как Aβ, фрагменты аполипопротеина E (ApoE) и α-синуклеин, могут повредить митохондрии, сказал Леннарт Маке из Институтов Гладстона и Калифорнийского университета в Сан-Франциско. В этом случае поврежденные митохондрии не будут основной причиной заболевания, а скорее будут вторичными по отношению к действиям агрегированных белков, которые и будут основной причиной. Значительное количество исследований показывает, что пептиды Aβ накапливаются в митохондриях, где они вызывают дисфункцию и апоптоз (Manczak et al., 2006; Yao et al., 2009).).
Одним из возможных механизмов токсичности белковых отложений для нейронов является нарушение аксонального транспорта митохондрий. Митохондрии генерируются в основном в теле клетки и должны активно транспортироваться в синапс, где потребность в энергии высока. Без митохондрий синаптическая функция может быть нарушена. Mucke и его команда оценили эффекты белков Aβ и tau на аксональный транспорт митохондрий (Vossel et al., 2010). Они обнаружили, что добавление олигомеров Aβ в культуру быстро ингибирует аксональный транспорт митохондрий в здоровых нейронах, что подтверждается более ранними исследованиями. Их также интересовало определение того, играет ли роль тау. Снижение уровня тау предотвратило индуцированное олигомером Aβ нарушение транспорта аксонов, не влияя на исходный аксональный транспорт. Полная элиминация тау путем нокдауна гена также имела тот же эффект. Они пришли к выводу, что «Aβ требует тау для нарушения аксонального транспорта, и что снижение уровня тау защищает от дефектов Aβ-индуцированного аксонального транспорта» (Vossel et al., 2010, стр. 19).8-а).
Mucke и его команда оценили эффекты белков Aβ и tau на аксональный транспорт митохондрий (Vossel et al., 2010). Они обнаружили, что добавление олигомеров Aβ в культуру быстро ингибирует аксональный транспорт митохондрий в здоровых нейронах, что подтверждается более ранними исследованиями. Их также интересовало определение того, играет ли роль тау. Снижение уровня тау предотвратило индуцированное олигомером Aβ нарушение транспорта аксонов, не влияя на исходный аксональный транспорт. Полная элиминация тау путем нокдауна гена также имела тот же эффект. Они пришли к выводу, что «Aβ требует тау для нарушения аксонального транспорта, и что снижение уровня тау защищает от дефектов Aβ-индуцированного аксонального транспорта» (Vossel et al., 2010, стр. 19).8-а).
Другим связанным с болезнью белком, который повреждает митохондрии, является ApoE. Ген ApoE является основным геном предрасположенности, идентифицированным для болезни Альцгеймера с поздним началом, и он находится на хромосоме 19. Нейроны продуцируют ApoE, когда они подвергаются стрессу под действием множества факторов, включая старение, окислительный стресс, травму и отложение белка. Считается, что синтез ApoE защищает нейроны от повреждений, а также восстанавливает и реконструирует их. Однако исследования показали, что продукты расщепления ApoE повреждают митохондрии (Brecht et al., 2004). По словам Макке, аллель ApoE e4, связанный с болезнью Альцгеймера, наиболее чувствителен к расщеплению, в то время как другие аллели ApoE менее чувствительны.
Нейроны продуцируют ApoE, когда они подвергаются стрессу под действием множества факторов, включая старение, окислительный стресс, травму и отложение белка. Считается, что синтез ApoE защищает нейроны от повреждений, а также восстанавливает и реконструирует их. Однако исследования показали, что продукты расщепления ApoE повреждают митохондрии (Brecht et al., 2004). По словам Макке, аллель ApoE e4, связанный с болезнью Альцгеймера, наиболее чувствителен к расщеплению, в то время как другие аллели ApoE менее чувствительны.
МИТОХОНДРИАЛЬНЫЕ ЗАБОЛЕВАНИЯ И ИХ ПОЛЕЗНОСТЬ ПРИ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЯХ
Учитывая неопределенность в отношении того, какую роль митохондриальная дисфункция играет в нейродегенеративных заболеваниях, Мута предположил ценность изучения первичных митохондриальных заболеваний, которые относятся к почти 150 генетическим заболеваниям, при которых поражение лежит в ген, кодирующий белок, непосредственно участвующий в биологии митохондрий. Болезни неоднородны, десятки находятся в центре внимания исследований на протяжении многих десятилетий. Вызванные генетическими мутациями или делециями одного гена, они следуют менделевскому или материнскому типу наследования.
Вызванные генетическими мутациями или делециями одного гена, они следуют менделевскому или материнскому типу наследования.
Митохондриальные заболевания могут пролить свет на нейродегенеративные заболевания, сказал Мутха, отчасти потому, что фенотипы заболеваний схожи. Например, некоторые фенотипы митохондриальных заболеваний включают атаксию, невропатию, миопатию, глухоту и слепоту. Действительно, несколько последующих презентаций были посвящены митохондриальной патологии при нейродегенеративных заболеваниях, таких как болезнь Паркинсона и БАС. Еще одна причина, по которой митохондриальные заболевания играют важную роль в нейродегенеративных заболеваниях, заключается в том, что, как и при нейродегенеративных заболеваниях, вовлекаются многие системы органов, а их генетика лучше охарактеризована благодаря амбициозному проекту, известному как Mitocarta, который представляет собой перечень более 1000 мышиных генов, кодирующих белки. которые локализуются в митохондриях (Pagliarini et al. , 2008). Наконец, митохондриальные заболевания ценны, по его мнению, тем, что они предоставляют «генетические крайности», которые могут помочь определить, может ли конкретное нейродегенеративное заболевание иметь митохондриальные дефекты в качестве основной причины. Мутха посоветовал искать связи между митохондриальными и нейродегенеративными заболеваниями, когда есть хотя бы какие-то точки соприкосновения, например, в патогенезе, патологии или биомаркерах.
, 2008). Наконец, митохондриальные заболевания ценны, по его мнению, тем, что они предоставляют «генетические крайности», которые могут помочь определить, может ли конкретное нейродегенеративное заболевание иметь митохондриальные дефекты в качестве основной причины. Мутха посоветовал искать связи между митохондриальными и нейродегенеративными заболеваниями, когда есть хотя бы какие-то точки соприкосновения, например, в патогенезе, патологии или биомаркерах.
Несмотря на то, что при микроскопии митохондрии выглядят одинаково, внешний вид обманчив. Мутха отметил огромную неоднородность митохондрий в разных тканях. Он сообщил, что после изучения 14 различных тканей исследования показали, что митохондрии из 2 разных тканей имеют только 75 процентов общих белков, тогда как остальные митохондриальные белки являются тканеспецифичными. Существует даже физиологическая неоднородность внутри отдельной клетки — митохондрии, например, могут иметь разные модели использования топлива. Учитывая разнообразие фенотипов и генотипов, Мутха выступал за системный подход к изучению митохондриальной функции. Такой подход сочетает в себе геномику, протеомику, метаболомику, биохимию и компьютерное моделирование, чтобы охватить динамический диапазон сложных взаимодействий внутри клеток и между тканями. Он заметил, что применение системной биологии к нейродегенеративным заболеваниям потребует определения составных частей, построения схем соединений для соединения этих частей, определения схем, вызывающих заболевание, и использования знаний для разработки методов лечения.
Такой подход сочетает в себе геномику, протеомику, метаболомику, биохимию и компьютерное моделирование, чтобы охватить динамический диапазон сложных взаимодействий внутри клеток и между тканями. Он заметил, что применение системной биологии к нейродегенеративным заболеваниям потребует определения составных частей, построения схем соединений для соединения этих частей, определения схем, вызывающих заболевание, и использования знаний для разработки методов лечения.
МИТОХОНДРИИ И СМЕРТЬ КЛЕТОК
Нейродегенеративные заболевания имеют общую черту: все они характеризуются высокой степенью гибели клеток. Здесь основное внимание уделяется митохондриям; митохондрии играют ключевую роль в регуляции гибели клеток, которая происходит в определенных областях мозга при всех нейродегенеративных заболеваниях. Гибель клеток бывает трех типов: (1) некроз, представляющий собой наиболее хаотичную форму гибели, которая включает набухание цитоплазмы, растворение ядра и лизис; (2) апоптоз, упорядоченная форма смерти, зависящая от АТФ, которая производит клеточные фрагменты, которые фагоцитирующие клетки способны поглощать и удалять до того, как содержимое клетки извергнется на окружающие клетки и вызовет повреждение; и (3) аутофагия, при которой клетка разрушает свою цитоплазму и органеллы с помощью лизосом (Martin et al. , 2010). Митохондрии — это места, где взаимодействуют антиапоптотические и проапоптотические белки, и они регулируют сигналы клеточной гибели.
, 2010). Митохондрии — это места, где взаимодействуют антиапоптотические и проапоптотические белки, и они регулируют сигналы клеточной гибели.
Ли Мартин из Университета Джона Хопкинса предупредил, что гибель клеток в моделях нейродегенеративных заболеваний у людей и животных может происходить по разным механизмам. Он сообщил о различиях между видами мышей и людей в факторах, контролирующих переход митохондриальной проницаемости (MPT), то есть увеличение проницаемости митохондриальных мембран для молекул с малой молекулярной массой. MPT возникает в результате открытия поры перехода митохондриальной проницаемости, белковой поры, образующейся в митохондриальных мембранах при определенных патологических состояниях. Индукция поры перехода проницаемости может привести к набуханию митохондрий и некрозу, а также играет важную роль в некоторых типах апоптоза. Мартин также отметил видовые различия в механизмах передачи сигналов каспаз, которые представляют собой ферменты, находящиеся под контролем митохондрий, которые имеют решающее значение для апоптоза, различия в субстратах касп, различия в механизмах слияния митохондрий и в механизмах передачи сигналов для репарации и метаболизма ДНК, среди прочего. Он заметил, что межвидовые различия в гибели клеток мешают переводу результатов, полученных с животных моделей, в клинические испытания на людях. Он предложил изменить дизайн доклинических исследований, чтобы меньше полагаться на мыши в качестве моделей и больше на нейроны, полученные из нервных стволовых клеток человека.
Он заметил, что межвидовые различия в гибели клеток мешают переводу результатов, полученных с животных моделей, в клинические испытания на людях. Он предложил изменить дизайн доклинических исследований, чтобы меньше полагаться на мыши в качестве моделей и больше на нейроны, полученные из нервных стволовых клеток человека.
ПОТЕНЦИАЛЬНЫЕ БИОМАРКЕРЫ И ТЕРАПИИ
Нет установленных биомаркеров или методов лечения для лечения митохондриальной дисфункции при нейродегенеративных заболеваниях. Уоллес сказал, что его лаборатория работает над их разработкой. Одним из разрабатываемых биомаркеров является спектроскопия кожи в ближнем инфракрасном диапазоне с использованием различных инфракрасных диодов, которые исследуют окислительно-восстановительный потенциал дыхательных цепей. Уоллес сказал, что его лаборатория также разрабатывает биомаркер с использованием анализа дыхания микроорганизмов. Они надеются получить некоторые суррогатные переменные, которые изменяются в режиме реального времени, а затем перейти к фазе I клинических испытаний и иметь по крайней мере показания безопасности/эффективности.
Что касается терапии, в этой главе уже упоминались некоторые из них, относящиеся к конкретным нейродегенеративным заболеваниям. Сосредоточившись вместо этого на родовых методах лечения митохондриальной дисфункции, Уоллес сказал, что его первоочередной задачей в терапии будет стимуляция образования большего количества митохондрий. Препараты для создания митохондрий тестируются на различных животных моделях и в системах клеточных культур. В частности, он отметил, что препарат безафибрат увеличивает биогенез митохондрий в раковых клетках и улучшает митохондриальную дисфункцию (Wang and Moraes, 2011). Он еще не был испытан на клетках головного мозга.
Другие варианты лечения, объяснил Уоллес, включают (1) каталитические антиоксиданты для защиты от АФК; (2) регуляторы внутриклеточного кальция; и (3) регуляторы окислительно-восстановительного потенциала через митохондриальную мембрану. Поддержание окислительно-восстановительного потенциала имеет решающее значение для целостности митохондрий и контроля окислительного фосфорилирования. Один участник отметил, что антиоксидантная терапия в клинических испытаниях всегда оказывалась неэффективной, но Уоллес ответил, что дозы, возможно, были недостаточно высокими. Другой участник посоветовал нацеливать митохондриальную терапию в случаях пороговых эффектов, то есть в момент, когда происходит значительное нарушение митохондриальной функции. Участник также отметил, что митохондриальная терапия может иметь вторичные недостатки.
Один участник отметил, что антиоксидантная терапия в клинических испытаниях всегда оказывалась неэффективной, но Уоллес ответил, что дозы, возможно, были недостаточно высокими. Другой участник посоветовал нацеливать митохондриальную терапию в случаях пороговых эффектов, то есть в момент, когда происходит значительное нарушение митохондриальной функции. Участник также отметил, что митохондриальная терапия может иметь вторичные недостатки.
ПОТРЕБНОСТИ В ИССЛЕДОВАНИЯХ И СЛЕДУЮЩИЕ ШАГИ, ПРЕДЛОЖЕННЫЕ ОТДЕЛЬНЫМИ УЧАСТНИКАМИ
Спикеры семинара определили множество вопросов для будущих исследований и другие возможности для будущих действий. Предложения, связанные с митохондриальной дисфункцией, собраны здесь, чтобы дать представление о диапазоне сделанных предложений. Предложения отождествляются с докладчиком, который их сделал, и не должны рассматриваться как отражение консенсуса на семинаре или одобрение Институтом медицины.
Развивать более глубокое понимание энергетической биологии и взаимодействия между биоэнергетикой и влиянием окружающей среды.
 (Уоллес)
(Уоллес)Определите биомаркеры для отслеживания функционирования митохондрий. (Ли, Мутха)
Найдите биомаркеры угасания митохондрий. (Mootha)
Определить новые методы лечения, повышающие биогенез митохондрий. (Wallace)
Определить методы лечения, препятствующие вкладу митохондрий в патогенез нейродегенеративного заболевания, включая методы лечения, которые усиливают митофагию или стимулируют активность PINK1 и паркина. (Мута, Юл)
Найдите методы лечения, которые детоксифицируют агрегаты мутантных белков, которые взаимодействуют с митохондриями. (Kowall)
Сноски
- 1
АФК образуются как побочный продукт окислительного фосфорилирования.
- 2
Примерно от 5 до 10 процентов случаев БАС являются семейными.
- 3
Деление митохондрий — это механизм контроля качества, при котором митохондрия делится на две части, одна здоровая, а другая содержит поврежденную часть митохондрий.