Гипофосфатазия у детей: Гипофосфатазия у детей. Три лица одной болезни | Бойков С.А., Черняк И.Ю., Шатохина Н.С., Гуркина Е.Ю., Бородина Н.А., Челабова Е.Ф., Эпоева С.А.

Что такое гипофосфатазия?
- Главная
- Пациентам
- Статьи
- Что такое гипофосфатазия?
Это редкое заболевание, при котором нарушается минерализация костей и зубов
Гипофосфатазия является редкой наследственной патологией, при которой нарушается развитие костей и зубов.
При этом расстройстве происходит нарушение минерализации – процесса усвоения костями и зубами различных минеральных веществ (в частности, кальция и фосфора), которые делают их твердыми и прочными.
Распространенность гипофосфатазии
Известно, что гипофосфатазия встречается редко. Ее распространенность изучена плохо, проведено мало исследований.
Согласно исследованию, основанному на записях в педиатрических клиниках США, с тяжелой гипофосфатазией рождается 1 из 100 000 детей. В докладе ученых отмечается, что, вероятно, более легкие формы заболевания встречаются гораздо чаще.
Более позднее исследование, опубликованное в журнале «Анналы генетики человека» (Annals of Human Genetics), указывает распространенность тяжелой гипофосфатазии среди европейцев около 1 на 300 000. Эти данные основаны на учете диагностированных случаев.
На основе генетических анализов исследователи указывают распространенность умеренных форм гипофосфатазии 1 на 6 730.
Причины и факторы риска гипофосфатазии
В основе развития гипофосфатазии лежит мутация гена ALPL, кодирующего щелочную фосфатазу, важный фермент, который расщепляет некоторые вещества, участвует в минерализации зубов и костей.
Мутация приводит к образованию дефектной щелочной фосфатазы, которая не может принимать участие в минерализации.
Без нормальной щелочной фосфатазы в организме начинают накапливаться различные соединения. Например, неорганический пирофосфат, который, как известно, подавляет процесс минерализации.
Гипофосфатазия может развиваться у ребенка, если оба родителя имеют дефектный ген.
Клинические формы и симптомы гипофосфатазии
Существует шесть клинических форм гипофосфатазии:
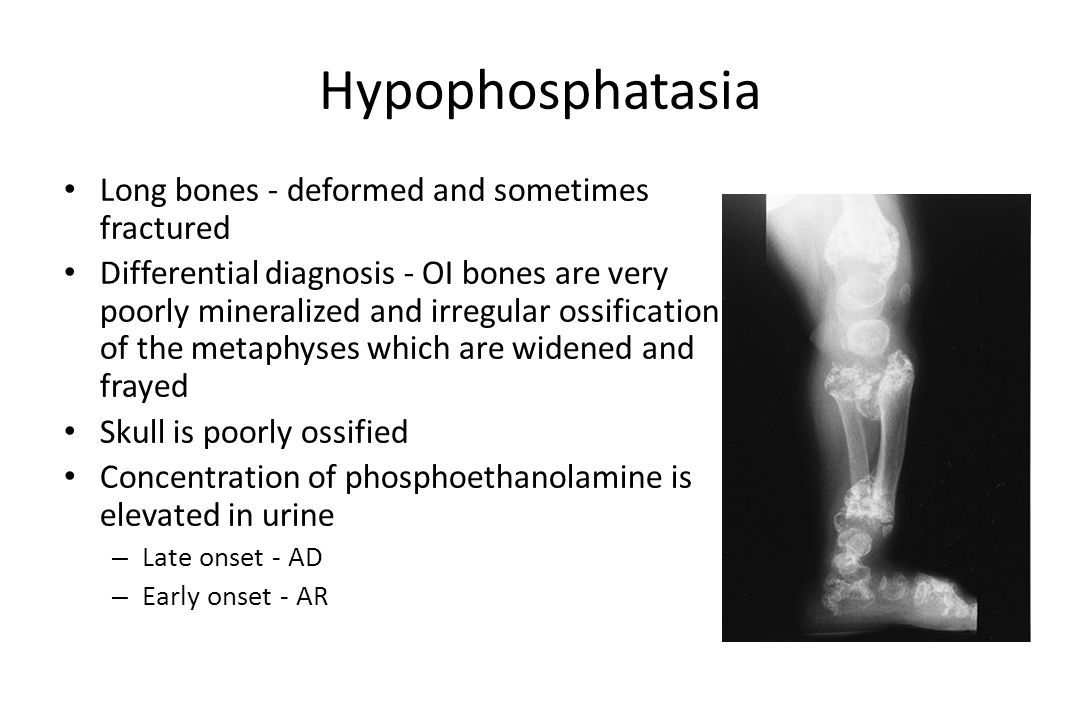
- Перинатальная несовместимая с жизнью форма. Такие дети рождаются с неокостеневшими «шпорами» на коже. У них отмечаются судороги, апноэ (остановка дыхания), укорочение длинных трубчатых костей. Больной ребенок может прожить несколько дней, но в конечном итоге погибает из-за нарушений дыхания, связанных с недоразвитием легких и грудной клетки.
- Доброкачественная перинатальная форма. Отмечается спонтанное улучшение состояния скелета плода, хотя изначально имелись нарушения. Как правило, улучшения состояния скелета и процесса минерализации происходят в третьем триместре беременности, но позже в течение жизни могут развиваться симптомы других форм гипофосфатазии.
- Инфантильная форма.
 Развивается в течение первых 6-ти месяцев жизни. У детей с этой формой заболевания возникают нарушения дыхания в результате рахита, а также отмечается повышение уровня кальция в крови, приводящее к ряду расстройств:
Развивается в течение первых 6-ти месяцев жизни. У детей с этой формой заболевания возникают нарушения дыхания в результате рахита, а также отмечается повышение уровня кальция в крови, приводящее к ряду расстройств:
- повышенная возбудимость;
- вялое сосание груди, снижение аппетита;
- рвота;
- снижение мышечного тонуса;
- жажда;
- частые мочеиспускания и склонность к обезвоживанию;
- запоры.
Около половины детей, страдающих инфантильной формой гипофосфатазии, погибают от дыхательных расстройств. Выжившие дети рано теряют молочные зубы, имеют низкий рост. - Ювенильная форма. Характеризуется целым рядом проблем со стороны зубов и костей, в том числе:
- рецидивирующие переломы и боли в костях;
- деформация, увеличение суставов;
- позднее начало ходьбы;
- невысокий рост;
- специфическая походка «вразвалку»;
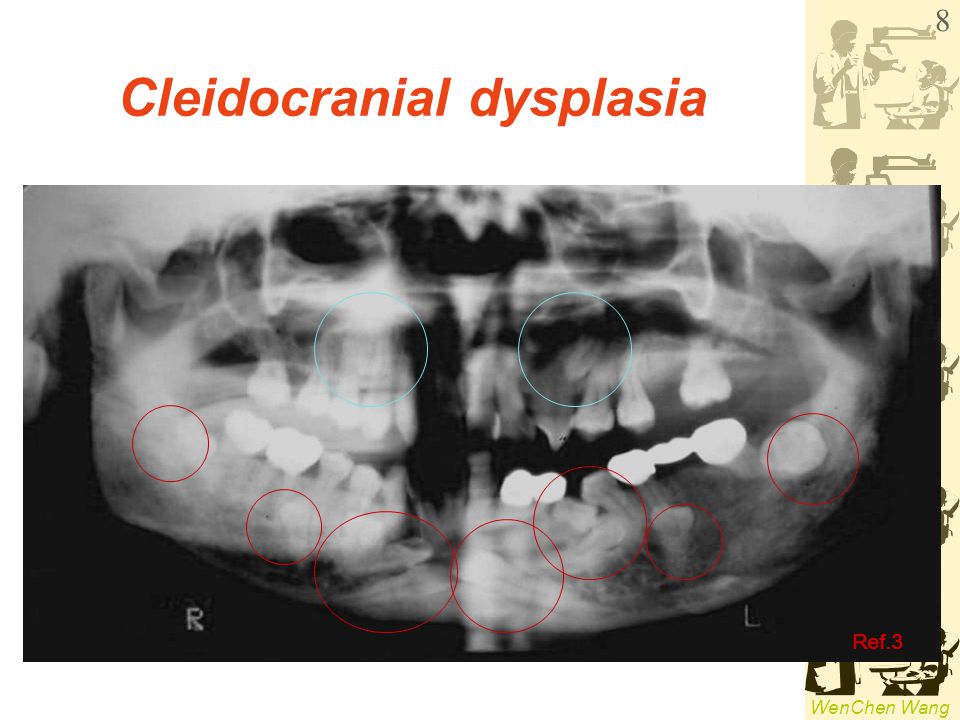
- преждевременное выпадение зубов, особенно резцов.

Если у вас возникла проблема, похожая на описанную в данной статье, обязательно обратитесь к нашим специалистам. Не ставьте диагноз самостоятельно!
Почему стоит позвонить нам сейчас:
- Ответим на все ваши вопросы за 3 минуты
- Бесплатная консультация
- Средний стаж работы врачей – 12 лет
- Удобство расположения клиник
Может наступить спонтанное выздоровление, но болезнь также может вернуться в более позднем возрасте. - Взрослая форма. Обычно развивается в среднем возрасте. Симптомы включают преждевременное выпадение зубов, боли в ногах из-за переломов напряжения. Эта форма заболевания не сокращает продолжительность жизни человека.
- Одонтогипофосфатазия. Обычно не приводит к проблемам с костями, как другие формы гипофосфатазии.
 Отмечаются нарушения со стороны зубов: сильный кариес, раннее выпадение молочных зубов.
Отмечаются нарушения со стороны зубов: сильный кариес, раннее выпадение молочных зубов.
Диагностика и лечение гипофосфатазии
Гипофосфатазию помогает обнаружить анализ крови на щелочную фосфатазу. Для подтверждения диагноза проводится исследование на генетические мутации.
Гипофосфатазия – неизлечимое заболевание. Лечение направлено на борьбу с симптомами и может включать следующие препараты и меры:
- Нестероидные противовоспалительные средства (НПВС) для борьбы с болью (особенно при ювенильной форме гипофосфатазии).
- Терипаратид – препарат, который используется для лечения остеопороза, а у взрослых пациентов с гипофосфатазией помогает бороться с переломами напряжения.
- Ограничение фосфатов в рационе питания, особенно у детей с задержкой роста.
- Физиотерапия.
- Регулярные осмотры стоматолога.
Также может быть полезна фермент-заместительная терапия, особенно для младенцев и детей, страдающих опасными для жизни формами гипофосфатазии.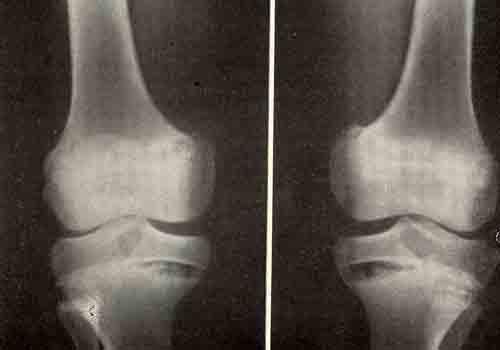
Инфантильная форма гипофосфатазии: клинический случай | Габрусская
1. Whyte MP. Hypophosphatasia — aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12(4):233–246. doi: 10.1038/nrendo.2016.
2. Whyte MP. Hypophosphatasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. Vol. 4. 8th ed. New York, NY: McGraw-Hill; 2001. Рр. 5313–5329.
3. Whyte MP, Zhang F, Wenkert D, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015; 75:229–239. doi: 10.1016/j.bone.2015.02.022.
4. Shohat M, Rimoin DL, Gruber HE, Lachman RS. Perinatal lethal hypophosphatasia; clinical, radiologic and morphologic findings. Pediatr Radiol. 1991;21(6):421–427. doi: 10.1007/bf02026677.
5. Silver MM, Vilos GA, Milne KJ. Pulmonary hypoplasia in neonatal hypophosphatasia. Pediatr Pathol. 1988;8(5):483–493. doi: 10.3109/15513818809022304.
Pediatr Pathol. 1988;8(5):483–493. doi: 10.3109/15513818809022304.
6. Fallon MD, Fallon MD, Teitelbaum SL, et al. Hypophosphatasia: clinicopathologic comparison of the infantile, childhood, and adult forms. Medicine (Baltimore). 1984;63(1):12–24.
7. Seshia SS, Derbyshire G, Haworth JC, Hoogstraten J. Myopathy with hypophosphatasia. Arch Dis Child. 1990;65(1):130–131. doi: 10.1136/adc.65.1.130.
8. Whyte MP, Teitelbaum SL, Murphy WA, et al. Adult hypophosphatasia: clinical, laboratory, and genetic investigation of a large kindred with review of the literature. Medicine (Baltimore). 1979;58(5):329–347.
9. Khandwala HM, Mumm S, Whyte MP. Low serum alkaline phosphatase activity and pathologic fracture: case report and brief review of hypophosphatasia diagnosed in adulthood. Endocr Pract. 2006;12(6):676–681. doi: 10.4158/EP.12.6.676.
10. Coe JD, Murphy WA, Whyte MP. Management of femoral fractures and pseudofractures in adult hypophosphatasia. J Bone Joint Surg Am. 1986;68(7):981–990.
1986;68(7):981–990.
11. Sobel EH., Clark LC, Fox RP, Robinow M. Rickets, deficiency of alkaline phosphatase activity and premature loss of teeth in childhood. Pediatrics. 1953;11(4):309–322.
12. Lundgren T, Westphal O, Bolme P, et al. Retrospective study of children with hypophosphatasia with reference to dental changes. Scand J Dent Res. 1991;99(5):357–364. doi: 10.1111/j.16000722.1991.tb01041.x.
13. Henthorn PS. Whyte MP. Missense mutations of the tissuenonspecific alkaline phosphatase gene in hypophosphatasia. Clin Chem. 1992;38(12):2501–2505.
14. Whyte MP. Hypophosphatasia. In: JP Bilezikian, LG Raisz, TJ Martin, 3rd ed. Principles of Bone Biology. Cambridge, MA, USA: Academic Press; 2008. Рp. 1573–1598.
15. Kozlowski K, Sutcliffe J, Barylak A, et al. Hypophosphatasia. Review of 24 cases. Pediatr Radiol. 1976;5(2):103–317. doi: 10.1007/bf00975316.
16. Whyte MP, Rockman-Greenberg C, Ozono K, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016;101(1):334–342. doi: 10.1210/jc.2015-3462.
J Clin Endocrinol Metab. 2016;101(1):334–342. doi: 10.1210/jc.2015-3462.
17. Kishnani PS, Rockman-Greenberg C, Rauch F, et al. Fiveyear efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone. 2019;121:149–162. doi: 10.1016/j.bone.2018.12.011.
18. Hogler W, Langman C, Gomes da Silva H, et al. Diagnostic delay is common among patients with hypophosphatasia: initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord. 2019;20(1):80. doi: 10.1186/s12891-019-2420-8.
19. Lee JY, So TY, Thackray J. A review on vitamin d deficiency treatment in pediatric patients. J Pediatr Pharmacol Ther. 2013; 18(4):277–291. doi: 10.5863/1551-6776-18.4.277.
20. Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. Horm Res Paediatr. 2016;85(2):83–106. doi: 10.1159/000443136.
21. Maman E, Borderie D, Roux C, Briot K. Absence of recognition of low alkaline phosphatase level in a tertiary care hospital. Osteoporos Int. 2016;27(3):1251–1254. doi: 10.1007/s00198-015-3346-0.
Osteoporos Int. 2016;27(3):1251–1254. doi: 10.1007/s00198-015-3346-0.
22. Deeb A, Elfatih A. Could alerting physicians for low alkaline phosphatase levels be helpful in early diagnosis of hypophosphatasia? J Clin Res Pediatr Endocrinol. 2018;10(1):19–24. doi: 10.4274/jcrpe.4426.
23. Gennero I, Conte-Auriol F, Salles JP. Laboratory diagnosis of hypophosphatasia. Arch Pediatr. 2017;24(5S2):5S57–5S60. doi: 10.1016/S0929-693X(18)30015-0.
24. Scriver CR, Cameron D. Pseudohypophosphatasia. N Engl J Med. 1969;281(11):604–606. doi: 10.1056/NEJM196909112811107.
25. Hofmann CE, Harmatz P, Vockley J, et al.; ENB-010-10 Study Group. Efficacy and safety of asfotase alfa in infants and young children with hypophosphatasia: a phase 2 open-label study. J Clin Endocrinol Metab. 2019;104(7):2735–2747. doi: 10.1210/jc.2018-02335.
26. Michigami T, Tachikawa K, Yamazaki M, et al. Hypophosphata sia in Japan: ALPL mutation analysis in 98 unrelated patients. Calcif Tissue Int. 2019. doi: 10.1007/s00223-019-00626-w.
doi: 10.1007/s00223-019-00626-w.
Гипофосфатазия у детей и подростков: клиника и лечение
. 2017 мая; 24 (5S2): 5S66-5S70.
doi: 10.1016/S0929-693X(18)30017-4.
А Ротенбюлер
1
, А Лингларт
2
Принадлежности
- 1 AP-HP, Справочный центр редких заболеваний метаболизма кальция и фосфатов, Платформа экспертизы редких заболеваний Париж-Юг, филиал OSCAR и педиатрическая служба эндокринологии, больница Бисетр-Париж-Юг, Ле-Кремлен-Бисетр, Франция ; INSERM U1169, Больница Бисетр, Кремль-Бисетр, Университет Париж-Сакле. Электронный адрес: [email protected].
- 2 AP-HP, Справочный центр редких заболеваний обмена кальция и фосфатов, Платформа экспертизы редких заболеваний Париж-Юг, филиал OSCAR и педиатрическая служба эндокринологии, больница Бисетр Париж-Юг, Кремль- Бисетр, Франция; ИНСЕРМ U1169, Больница Бисетр, Кремль-Бисетр, Университет Париж-Сакле.

PMID:
29405935
DOI:
10.1016/S0929-693X(18)30017-4
А. Ротенбюлер и соавт.
Арка Педиатр.
2017 май.
. 2017 мая; 24 (5S2): 5S66-5S70.
doi: 10.1016/S0929-693X(18)30017-4.
Авторы
А Ротенбюлер
1
, А Лингларт
2
Принадлежности
- 1 AP-HP, Справочный центр редких заболеваний обмена кальция и фосфатов, Платформа экспертизы редких заболеваний Париж-Юг, отделение OSCAR и педиатрическая служба эндокринологии, больница Бисетр Париж-Юг, Ле Кремль-Бисетр, Франция; ИНСЕРМ U1169, Больница Бисетр, Кремль-Бисетр, Университет Париж-Сакле.
 Электронный адрес: [email protected].
Электронный адрес: [email protected]. - 2 AP-HP, Справочный центр редких заболеваний обмена кальция и фосфатов, Платформа экспертизы редких заболеваний Париж-Юг, филиал OSCAR и педиатрическая служба эндокринологии, больница Бисетр Париж-Юг, Кремль- Бисетр, Франция; INSERM U1169, Больница Бисетр, Кремль-Бисетр, Университет Париж-Сакле.
PMID:
29405935
DOI:
10.1016/S0929-693X(18)30017-4
Абстрактный
Гипофосфатазия (HPP) является редким генетическим заболеванием из-за мутаций потери функции в гене, кодирующем щелочную фосфатазу печени (ALPL), который кодирует тканевую неспецифическую щелочную фосфатазу (TNSALP) или ALP. Ювенильный HPP, по определению, диагностируется в возрасте от 6 месяцев до зрелого возраста. Клинические признаки и симптомы ювенильного ГПП весьма разнообразны по своей картине, степени тяжести и течению. Наиболее известны костные (нарушение минерализации костей, деформации ног, боли, рахит, аномалии роста) и зубные (преждевременная потеря молочных зубов) аномалии. Однако у подростков часто преобладают мышечные и суставные аномалии. Варианты лечения в настоящее время остаются ограниченными симптоматическим лечением боли и нарушения функции. Обнадеживающие результаты ферментозаместительной терапии были продемонстрированы у детей с тяжелой формой ГПП. Эффективность и долгосрочные преимущества у пациентов с ювенильной формой еще предстоит доказать.
Ювенильный HPP, по определению, диагностируется в возрасте от 6 месяцев до зрелого возраста. Клинические признаки и симптомы ювенильного ГПП весьма разнообразны по своей картине, степени тяжести и течению. Наиболее известны костные (нарушение минерализации костей, деформации ног, боли, рахит, аномалии роста) и зубные (преждевременная потеря молочных зубов) аномалии. Однако у подростков часто преобладают мышечные и суставные аномалии. Варианты лечения в настоящее время остаются ограниченными симптоматическим лечением боли и нарушения функции. Обнадеживающие результаты ферментозаместительной терапии были продемонстрированы у детей с тяжелой формой ГПП. Эффективность и долгосрочные преимущества у пациентов с ювенильной формой еще предстоит доказать.
© 2017 Эльзевир Массон САС. Все права защищены.
Похожие статьи
Заместительная терапия щелочной фосфатазой при гипофосфатазии в разработке и практике.

Боуден С.А., Фостер Б.Л.
Боуден С.А. и др.
Adv Exp Med Biol. 2019;1148:279-322. дои: 10.1007/978-981-13-7709-9_13.
Adv Exp Med Biol. 2019.PMID: 31482504
Обзор.
Перинатальная и инфантильная гипофосфатазия: клиника и лечение.
Баужат Г., Мишо С., Ле Куан Санг К.Х., Кормье-Дэр В.
Баужат Г. и соавт.
Арка Педиатр. 2017 мая; 24 (5S2): 5S61-5S65. doi: 10.1016/S0929-693X(18)30016-2.
Арка Педиатр. 2017.PMID: 29405934
Гипофосфатазия: обзор за 2017 год.
Уайт, член парламента.
Уайт депутат.
Кость. 2017 сен;102:15-25. doi: 10.1016/j.bone.2017.02.011. Epub 2017 24 февраля.
Кость. 2017.PMID: 28238808
Обзор.
Гипофосфатазия: биохимические признаки подтверждают расширенную педиатрическую клиническую нозологию.
Уайт М.П., Коберн С.П., Райан Л.М., Эриксон К.Л., Чжан Ф.
Уайт М.П. и др.
Кость. 2018 Май; 110:96-106. doi: 10.1016/j.bone.2018.01.022. Epub 2018 31 января.
Кость. 2018.PMID: 29360619
Гипофосфатазия: от диагностики к лечению.
Саймон С., Реш Х., Клаусхофер К., Рошгер П., Зверина Дж., Косьян Р.
Саймон С. и др.
Curr Rheumatol Rep. 2018 Sep 10;20(11):69. doi: 10.1007/s11926-018-0778-5.
Curr Rheumatol Rep. 2018.PMID: 30203264
Обзор.
Посмотреть все похожие статьи
Цитируется
Гипофосфатазия.
Турнис С., Явропулу М.П., Полизос С.А., Доулгераки А.
Турнис С. и др.
Дж. Клин Мед. 2021 1 декабря; 10 (23): 5676. doi: 10.3390/jcm10235676.
doi: 10.3390/jcm10235676.
Дж. Клин Мед. 2021.PMID: 34884378
Бесплатная статья ЧВК.Обзор.
термины MeSH
вещества
Дополнительные понятия
NORD (Национальная организация по редким заболеваниям)
Гипофосфатазия
NORD выражает благодарность Майклу П. Уайту, доктору медицинских наук, научно-медицинскому директору Центра метаболических заболеваний костей и молекулярных исследований Детской больницы Шрайнерс в Сент-Луисе. Луи и профессор медицины, педиатрии и генетики, отделение костных и минеральных заболеваний, Медицинская школа Вашингтонского университета; Сент-Луис, штат Миссури, США, за помощь в подготовке этого отчета.
Synonyms of Hypophosphatasia
- HPP
- Rathbun disease
Subdivisions of Hypophosphatasia
- hypophosphatasia, perinatal
- hypophosphatasia, infantile
- hypophosphatasia, childhood (mild versus severe)
- hypophosphatasia, adult
- odontohypophosphatasia
- псевдогипофосфатазия
Признаки и симптомы
Причины
HPP вызывается мутациями в ALPL ген.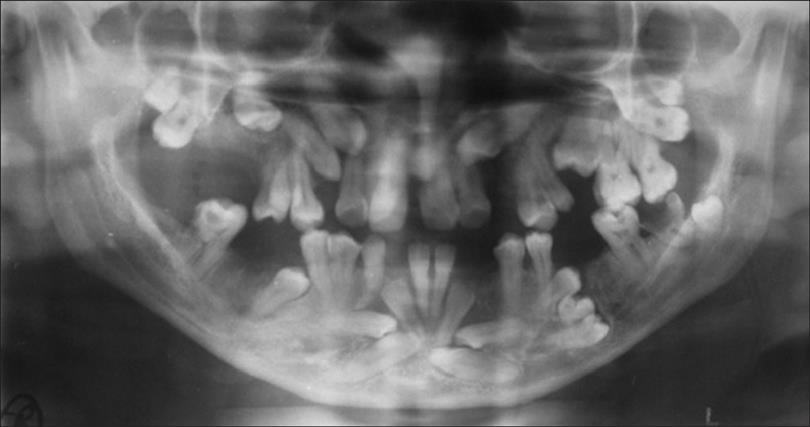 Это единственный ген, вызывающий HPP. Гены предоставляют инструкции для создания белков, которые выполняют важную функцию в организме. Когда происходит мутация, белок может быть неисправным, неэффективным или отсутствовать, как в HPP. В зависимости от функции белка может быть нарушена одна или несколько систем органов тела.
Это единственный ген, вызывающий HPP. Гены предоставляют инструкции для создания белков, которые выполняют важную функцию в организме. Когда происходит мутация, белок может быть неисправным, неэффективным или отсутствовать, как в HPP. В зависимости от функции белка может быть нарушена одна или несколько систем органов тела.
Ген ALPL создает (кодирует) тип белка, называемый ферментом TNSALP. Ферменты — это специализированные белки, которые расщепляют определенные химические вещества в организме. TNSALP необходим для правильного развития и здоровья костей и зубов, его много в скелете, печени и почках. Мутации в 9Ген 0161 ALPL снижает активность TNSALP, что, в свою очередь, приводит к накоплению фосфоэтаноламина (ФЭА), пиридоксаль-5′-фосфата (PLP) и неорганического пирофосфата (PPi). Неорганический пирофосфат является ингибитором минерализации, который контролирует поступление минералов в скелет. Повышенные уровни PPi могут блокировать попадание кальция и фосфора в кости и тем самым вызывать повышенный уровень кальция в крови и моче. Как правило, снижение активности фермента TNSALP коррелирует с тяжестью HPP (меньшая активность фермента вызывает более тяжелое заболевание).
Как правило, снижение активности фермента TNSALP коррелирует с тяжестью HPP (меньшая активность фермента вызывает более тяжелое заболевание).
HPP может наследоваться по аутосомно-рецессивному (поражающему братьев и сестер) или аутосомно-доминантному (поражающему несколько поколений) типу. Перинатальная и инфантильная формы ГПП наследуются по аутосомно-рецессивному типу. Детская форма может быть как аутосомно-рецессивной, так и аутосомно-доминантной. Взрослая форма и одонтогипофосфатазия обычно являются аутосомно-доминантными заболеваниями, но редко аутосомно-рецессивными.
Доминантные генетические расстройства возникают, когда для возникновения определенного заболевания необходима только одна копия неработающего гена. Неработающий ген может быть унаследован от любого из родителей или может быть результатом измененного (мутированного) гена у больного человека. Риск передачи неработающего гена от больного родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Рецессивные генетические нарушения возникают, когда человек наследует нерабочий ген от каждого родителя. Если человек получает один рабочий ген и один неработающий ген болезни, он будет носителем болезни, но обычно не будет проявлять симптомов. Риск для двух родителей-носителей передать неработающий ген и, следовательно, родить больного ребенка составляет 25% при каждой беременности. Риск рождения ребенка-носителя, как и у родителей, составляет 50% при каждой беременности. Вероятность того, что ребенок получит рабочие гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
Пораженное население
HPP поражает мужчин и женщин в равной степени. По оценкам, в Канаде тяжелый HPP встречается примерно у 1 из 100 000 живорождений. Общая заболеваемость и распространенность различных форм ГПП плохо изучены или неизвестны. Более легкие случаи могут остаться недиагностированными или неправильно диагностированными. HPP встречается с наибольшей частотой у меннонитов в Канаде, относительно распространен в Японии и, по-видимому, редко встречается у лиц чернокожего происхождения.
Диагностика
HPP диагностируется путем выявления его симптомов и осложнений, начиная с подробного анамнеза пациента. Признаки ГПП выявляются при тщательном клиническом обследовании и подтверждаются рутинными рентгенологическими исследованиями и различными лабораторными исследованиями, включая биохимические исследования. Врачам часто легко определить HPP для тех, кто знаком с этим заболеванием или имеет опыт его лечения. Понятно, однако, что большинство врачей мало или совсем не знают о HPP. Следовательно, пострадавшие лица и семьи могут столкнуться с досадной задержкой в диагностике. Теперь анализ генетических мутаций Ген ALPL доступен в коммерческих лабораториях для подтверждения диагноза HPP.
Клиническое обследование и обследование
Иногда впервые подозрение на HPP возникает при рутинном анализе крови, включающем анализ щелочной фосфатазы (ЩФ). В противном случае признаки и симптомы обычно приводят к рутинному тестированию, а низкий уровень ЩФ выделяется и распознается. Лица с HPP имеют сниженную активность ALP в сыворотке для своего возраста, за исключением крайне редких случаев нормального уровня ALP при псевдогипофосфатазии. Выявление недостаточной активности ALP согласуется с HPP, но не является диагностическим, поскольку причиной может быть множество других состояний. Кроме того, некоторые люди, которые являются генетическими «носителями» HPP, но у которых не проявляются какие-либо симптомы, также могут иметь низкий уровень ЩФ в крови.
Лица с HPP имеют сниженную активность ALP в сыворотке для своего возраста, за исключением крайне редких случаев нормального уровня ALP при псевдогипофосфатазии. Выявление недостаточной активности ALP согласуется с HPP, но не является диагностическим, поскольку причиной может быть множество других состояний. Кроме того, некоторые люди, которые являются генетическими «носителями» HPP, но у которых не проявляются какие-либо симптомы, также могут иметь низкий уровень ЩФ в крови.
Важно отметить, что активность ALP в сыворотке зависит от возраста. Здоровые дети обычно имеют более высокий уровень ЩФ, чем здоровые взрослые. Если лаборатория, выполняющая тестирование, дает только нормальный диапазон для взрослых, диагноз ГПП у ребенка может быть пропущен, поскольку его/ее активность ЩФ будет ошибочно считаться «нормальной».
В США и других странах диагноз HPP может быть подтвержден, но не поставлен путем измерения уровня в крови формы витамина B6, называемой пиридоксаль-5′-фосфатом (PLP). Этот тест проводится несколькими коммерческими лабораториями. Люди с HPP имеют повышенные уровни, потому что PLP обычно расщепляется TNSALP. PLP повышен даже при легком HPP. Однако некоторые генетические носители HPP могут иметь повышенный PLP. Кровь и моча могут быть проверены на повышенное количество фосфоэтаноламина (ФЭА), другого химического вещества, обычно расщепляемого TNSALP, однако у некоторых людей с HPP уровни ФЭА в норме, а повышение ФЭА может наблюдаться при других метаболических заболеваниях костей. Скрининг на повышенный PLP предпочтительнее скрининга на повышенный PEA, поскольку он более чувствителен, точен и дешевле.
Этот тест проводится несколькими коммерческими лабораториями. Люди с HPP имеют повышенные уровни, потому что PLP обычно расщепляется TNSALP. PLP повышен даже при легком HPP. Однако некоторые генетические носители HPP могут иметь повышенный PLP. Кровь и моча могут быть проверены на повышенное количество фосфоэтаноламина (ФЭА), другого химического вещества, обычно расщепляемого TNSALP, однако у некоторых людей с HPP уровни ФЭА в норме, а повышение ФЭА может наблюдаться при других метаболических заболеваниях костей. Скрининг на повышенный PLP предпочтительнее скрининга на повышенный PEA, поскольку он более чувствителен, точен и дешевле.
При тяжелом ГПП, особенно перинатальной и младенческой формах, рентгенологическое исследование костей позволяет выявить диагностические изменения ГПП. Однако эти изменения могут не распознаваться как ГПП, за исключением радиологов, знакомых с ГПП.
Молекулярно-генетическое тестирование может подтвердить диагноз HPP, поскольку оно позволяет обнаружить мутации в гене ALPL , вызывающем HPP, но оно доступно только в качестве диагностической услуги в определенных лабораториях.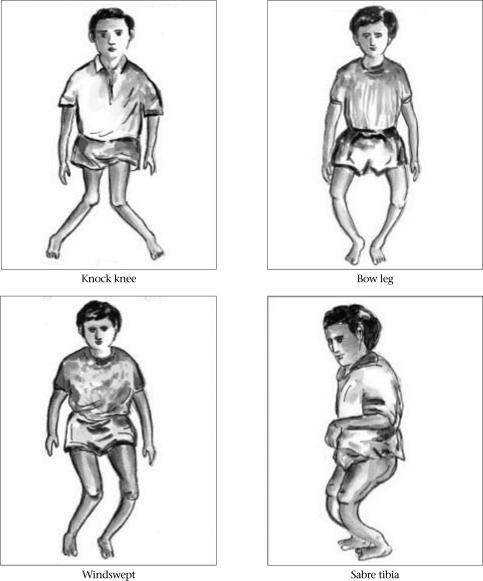 Тест может быть дорогим и часто не требуется для диагностики HPP.
Тест может быть дорогим и часто не требуется для диагностики HPP.
Standard Therapies
В 2015 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило асфотазу альфа (Strensiq) в качестве первого лекарственного препарата для лечения ГПП в перинатальном, младенческом и юношеском возрасте. В США пациенты любого возраста с ГПП в детском возрасте имеют право на эту заместительную терапию TNSALP, направленную на костную ткань, вводимую подкожной инъекцией.
Поддерживающее лечение HPP направлено на конкретные симптомы и осложнения. Для лечения может потребоваться команда специалистов. Для комплексного лечения могут потребоваться педиатры, хирурги-ортопеды, педодонтологи, специалисты по обезболиванию и другие медицинские работники.
Нестероидные противовоспалительные препараты (НПВП) могут уменьшить боль в костях и суставах. НПВП требуют осторожности и наблюдения за побочными эффектами (например, они могут повредить желудок и почки), особенно в избытке и при слишком длительном применении.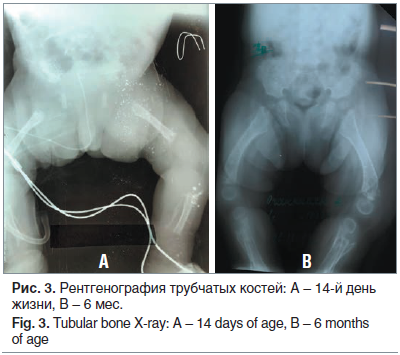 Если краниосиностоз вызывает внутричерепное давление, может потребоваться шунтирование или операция на черепе.
Если краниосиностоз вызывает внутричерепное давление, может потребоваться шунтирование или операция на черепе.
Витамин B6 может помочь контролировать специфические судороги у детей с тяжелыми заболеваниями. Людям с повышенным уровнем кальция в крови (гиперкальциемия) может потребоваться ограничение потребления кальция с пищей, гидратация, прием некоторых диуретиков и, возможно, инъекций кальцитонина.
Рекомендуется как можно раньше начать регулярный уход за зубами. Физиотерапия и трудотерапия могут быть полезными.
Взрослым с рецидивирующими переломами длинных костей может потребоваться ортопедический «стержень», при котором металлический стержень вставляется в центральную полость длинных костей для повышения стабильности и прочности. Специальные медицинские устройства (ортопедические стельки) могут помочь при переломах стопы (плюсневых костей).
Пациентам следует избегать бисфосфонатов, класса препаратов, используемых для лечения других заболеваний костей, таких как остеопороз.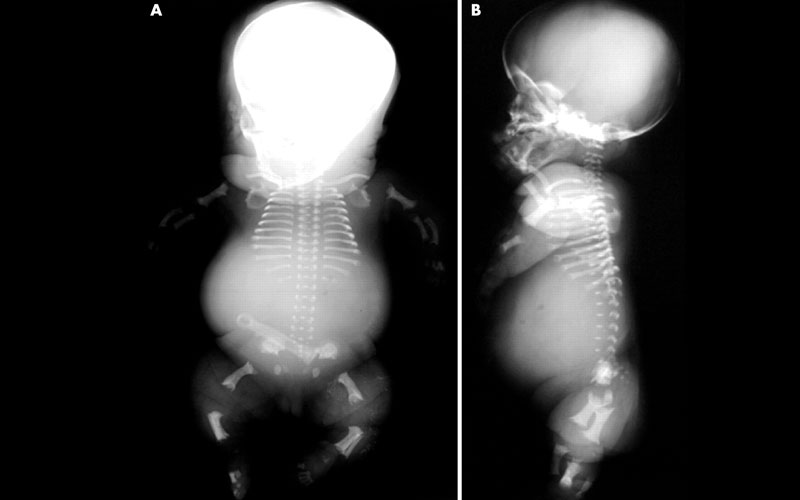 Бисфосфонаты могут ухудшить ГПН или вызвать проблемы у лиц с невыявленным ГПН. Примерами бисфосфонатных препаратов являются алендронат, ибандронат, памидронат, ризедронат и золендронат.
Бисфосфонаты могут ухудшить ГПН или вызвать проблемы у лиц с невыявленным ГПН. Примерами бисфосфонатных препаратов являются алендронат, ибандронат, памидронат, ризедронат и золендронат.
Генетическое консультирование может быть полезным для больных людей и их семей. Для детей с HPP психосоциальная поддержка для всей семьи может быть полезной.
Исследовательская терапия
Некоторые виды гормона паращитовидной железы, такие как терипаратид, вводились «не по прямому назначению» нескольким взрослым с ГПП, осложненным стрессовыми переломами плюсневых костей или псевдопереломами бедренной кости, что привело к заживлению переломов. Препарат не разрешен к применению у детей. Необходимы дополнительные исследования, чтобы определить долгосрочную безопасность и эффективность терипаратида при лечении HPP.
Трансплантация клеток костного мозга использовалась для лечения тяжелого HPP. Один пациент также получил костные фрагменты и культивированные остеобласты, костеобразующие клетки.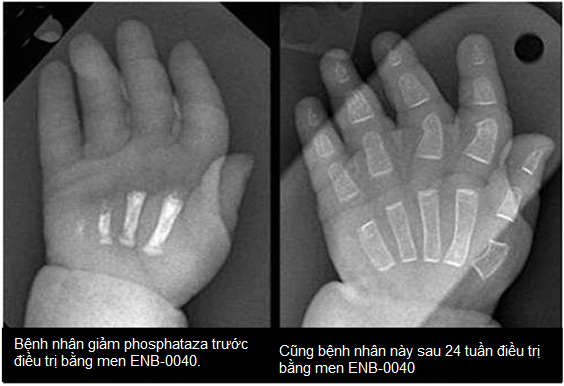
Сообщалось также о предварительных краткосрочных результатах применения для ГПП антител против склеростина. Склеростин — это белок, обнаруженный в клетках, встроенных в кость, известных как остеоциты. Склеростин помогает уменьшить (подавить) остеобласты. Было показано, что антитела, действующие против склеростина, увеличивают костную массу при остеопорозе.
Для получения дополнительной информации о HPP обращайтесь:
Майкл П. Уайт, MD
Медико-научный директор
Центр метаболических заболеваний костей и молекулярных исследований Shriners Hospitals for Children-St. Луи;
4400 Clayton Ave
Сент-Луис, Миссури 63110
Эл. исследует более 100 редких заболеваний костей. Центр известен во всем мире своим опытом лечения нескольких редких заболеваний, включая HPP. Исследовательская группа провела клинические испытания нового лечения HPP (асфотаза альфа), и они наблюдают за большим количеством детей и взрослых пациентов с HPP, чем где-либо еще в мире.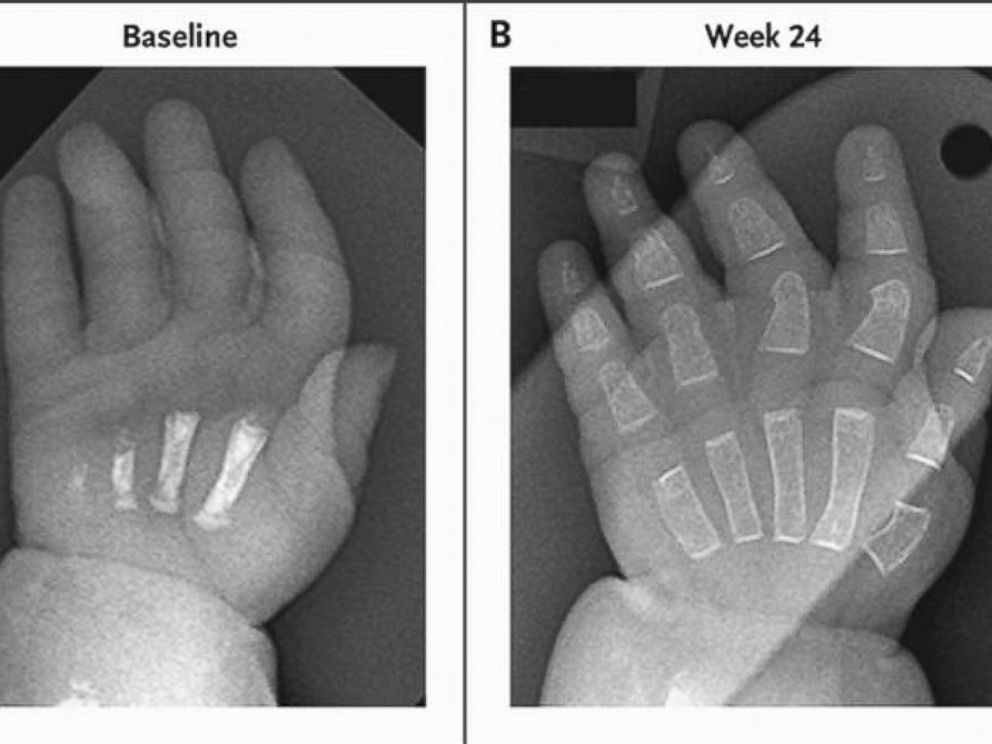 Центр служит глобальным ресурсом для пациентов и врачей, которые ищут информацию о редких генетических заболеваниях костей, таких как HPP.
Центр служит глобальным ресурсом для пациентов и врачей, которые ищут информацию о редких генетических заболеваниях костей, таких как HPP.
Информация о текущих клинических испытаниях размещена в Интернете на сайте www.clinicaltrials.gov. Все исследования, финансируемые правительством США, а некоторые из них поддерживаются частным сектором, публикуются на этом правительственном веб-сайте.
Для получения информации о клинических испытаниях, проводимых в Клиническом центре NIH в Бетесде, штат Мэриленд, обращайтесь в отдел набора пациентов NIH:
Бесплатный звонок: (800) 411-1222
Телетайп: (866) 411-1010
Электронная почта: [email protected]
Некоторые текущие клинические испытания также размещены на следующей странице веб-сайта NORD:
https://rarediseases.org/for-patients-and-families/information-resources/info-clinical-trials-and-research-studies/
Для получения информации о клинических испытаниях, спонсируемых частными источниками, в основном, обращайтесь :www.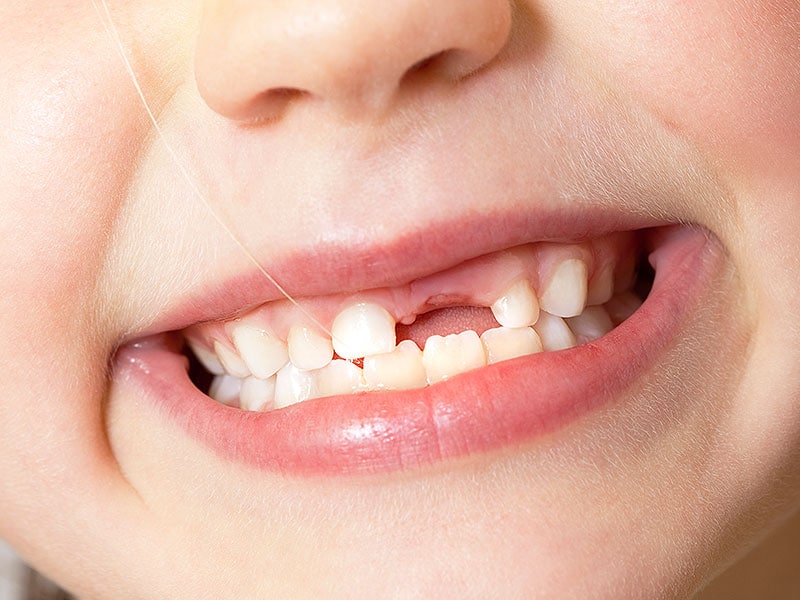 centerwatch.com
centerwatch.com
Для получения дополнительной информации о клинических испытаниях, проведенных в Европе, обращайтесь: https://www.clinicaltrialsregister.eu/
Ссылки
УЧЕБНИКИ
Whyte MP. Гипофосфатазия и как щелочная фосфатаза способствует минерализации. Глава 28 В: Генетика биологии костей и заболеваний скелета (2-е изд.). Таккер Р.В., Уайт М.П., Эйсман Дж., Игараши Т., ред. 2018 Elsevier/Academic Press, Лондон, Великобритания.
Hu JCC, Simmer JP. Гипофосфатазия. В: Руководство NORD по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003:317-318.
Горлин Р.Дж., Коэн М.М.Джр, Хеннекам Р.К.М. ред. Синдромы головы и шеи. 4-е изд. Издательство Оксфордского университета, Нью-Йорк, штат Нью-Йорк; 2001: 161-164.
ОБЗОРНЫЕ СТАТЬИ
Уайт MP: Гипофосфатазия: ферментозаместительная терапия открывает новые возможности и новые проблемы (перспектива). Журнал исследований костей и минералов 2017; 32: 667-675.
Уайт MP: Гипофосфатазия: этиология, нозология, патогенез, диагностика и лечение.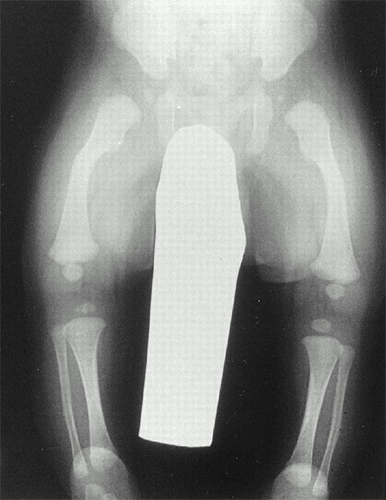 Nature Reviews Endocrinology 2016; 12: 233-46.
Nature Reviews Endocrinology 2016; 12: 233-46.
Миллан Дж.Л., Уайт М.П.: Щелочная фосфатаза и гипофосфатазия. Calcified Tissue International 2016; 98:398-416.
Озоно К. Ферментозаместительная терапия гипофосфатазии. Клин Кальций. 2014;24:257-263. http://www.ncbi.nlm.nih.gov/pubmed/24473359
Уайт, член парламента. Физиологическая роль щелочной фосфатазы исследована при гипофосфатазии. Энн NY Acad Sci. 2010;1192:190-200. http://www.ncbi.nlm.nih.gov/pubmed/20392236
СТАТЬИ В ЖУРНАЛЕ
Whyte MP, Zhang F, Wenkert D, Mumm S, Berndt TJ, Kumar R. Гиперфосфатемия с низким уровнем FGF7 и нормальным уровнем FGF23 и sFRP4 в циркуляции характеризует детскую гипофосфатазию. Кость 2020; 134:115300.
Уайт М.П., Макалистер В.Х., Мамм С., Бирхалс А.Дж. Отсутствие кальцификации сосудов на компьютерной томографии сердца при лечении асфотазой альфа у пожилой женщины с гипофосфатазией. Кость 2019; 122: 231-236.
Уайт М.П., Кобурн С.П., Райан Л.М., Эриксон К. Л., Чжан Ф. Гипофосфатазия: биохимические признаки подтверждают расширенную педиатрическую клиническую нозологию. Кость 2018; 110: 96-106.
Л., Чжан Ф. Гипофосфатазия: биохимические признаки подтверждают расширенную педиатрическую клиническую нозологию. Кость 2018; 110: 96-106.
Камачо PM, Mazhari1 AM, Wilczynski1 C, Kadanoff R, Mumm S, Whyte MP. Гипофосфатазия у взрослых, получавшая лечение терипаратидом: отчет о 2 пациентах и обзор литературы. Эндокринная практика 2016; 22: 941-50.
Уайт М.П., Мэдсон К.Л., Филлипс Д., Ривз А., Макалистер В.Х., Якимоски А., Мак К., Гамильтон К., Каган К., Мелиан А., Томпсон Д., Мозли С., Одрлин Т., Гринберг Ч.Р. Асфотазо-альфа-терапия у детей с гипофосфатазией. JCI Insight 2016; e85971;1- 10.
Whyte MP, Greenber CR, Ozone K, Riese R, Moseley S, Melian A, Thompson D, Hofmann C: Лечение асфотазой альфа улучшает выживаемость при пренатальной и детской гипофосфатазии. Journal of Clinical Endocrinology and Metabolism 2016; 101: 334-42.
Уайт MP, Mumm S, McAlister WH, Mack K, Benigno M, Kempa LG, Franken A, Lim VT, Ericson KL, Coburn SP, Ryan LM, Wenkert D, Zhang F: Гипофосфатазия: исследование естественного течения 101 больного ребенка, изученное в одном исследовательском центре. Кость 2016;93:125-138.
Кость 2016;93:125-138.
Whyte MP, Zhang F, Wenkert D, McAlister WH, Mack KE, Benigno MC, Coburn SP, Wagy S, Griffin DM, Ericson KL, Mumm S: Гипофосфатазия: валидация и расширение клинической нозологии для детей с 25-летним стажем с 173 педиатрическими пациентами. Кость 2015;75:229-39.
McKiernan FE, Berg RL, Fuehrer J. Клинические и рентгенологические данные у взрослых с персистирующей гипофосфатаземией. Джей Боун Шахтер Рез. 2014;29:1651-1660. http://www.ncbi.nlm.nih.gov/pubmed/24443354
Такетани Т., Онигата К., Кобаяши Х. и др. Клинические и генетические аспекты гипофосфатазии у японских пациентов. Арч Дис Чайлд. 2014;99:211-215. http://www.ncbi.nlm.nih.gov/pubmed/24276437
Guañabens N, Mumm S, Möller I, et al. Кальцифицирующий периартрит как единственное клиническое проявление гипофосфатазии у сестер среднего возраста. Джей Боун Шахтер Рез. 2014;29:929-934. http://www.ncbi.nlm.nih.gov/pubmed/24123110
Matsushita M, Kitoh H, Michigami T, Tachikawa K, Ishiguro N. Доброкачественная пренатальная гипофосфатазия: излечимое заболевание, которое нельзя упустить. Педиатр Радиол. 2014;44:340-343. http://www.ncbi.nlm.nih.gov/pubmed/24145968
Доброкачественная пренатальная гипофосфатазия: излечимое заболевание, которое нельзя упустить. Педиатр Радиол. 2014;44:340-343. http://www.ncbi.nlm.nih.gov/pubmed/24145968
Уайт М.П., Лилаваттана Р., Рейнус В.Р. и др. Острая тяжелая гиперкальциемия после травматических переломов и иммобилизации при гипофосфатазии, осложненной хронической почечной недостаточностью. J Clin Endocrinol Metab. 2013;98:4606-4612. http://www.ncbi.nlm.nih.gov/pubmed/24064686
Berkseth KE, Tebben PJ, Drake MT, et al. Клинический спектр гипофосфатазии, диагностированной у взрослых. Кость. 2013;54:21-27. http://www.ncbi.nlm.nih.gov/pubmed/23352924
Whyte MP, Greenberg CR, Salman NJ, et al. Ферментозаместительная терапия при жизнеугрожающей гипофосфатазии. N Engl J Med. 2012;366:904-913. http://www.ncbi.nlm.nih.gov/pubmed/22397652
Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP. «Атипичные переломы бедренной кости» при воздействии бисфосфонатов при гипофосфатазии у взрослых. Джей Боун Шахтер Рез.
2012;27:987-994. http://www.ncbi.nlm.nih.gov/pubmed/22322541
Wenkert D, McAlister WH, Coburn SP, et al. Гипофосфатазия: несмертельное заболевание, несмотря на скелетное предлежание в утробе матери (17 новых случаев и обзор литературы). Джей Боун Шахтер Рез. 2011;26:2389-2398. http://www.ncbi.nlm.nih.gov/pubmed/21713987
Stevenson DA, Carey JC, Coburn SP, et al. Аутосомно-рецессивная гипофосфатазия, проявляющаяся внутриутробно деформацией длинных костей, но демонстрирующая спонтанное постнатальное улучшение. J Clin Endocrinol Metab. 2008;93:3443-3448. http://www.ncbi.nlm.nih.gov/pubmed/18559907
Whyte MP, Mumm S, Deal C. Гипофосфатазия у взрослых, лечение терипаратидом. J Clin Endocrinol Metab. 2007;92:1203-1208. http://www.ncbi.nlm.nih.gov/pubmed/17213282
Cahill RA, Wenkert D, Perlman SA, et al. Детская гипофосфатазия: испытание трансплантационной терапии с использованием костных фрагментов и культивируемых остеобластов. J Clin Endocrinol Metab. 2007;92:2923-2930.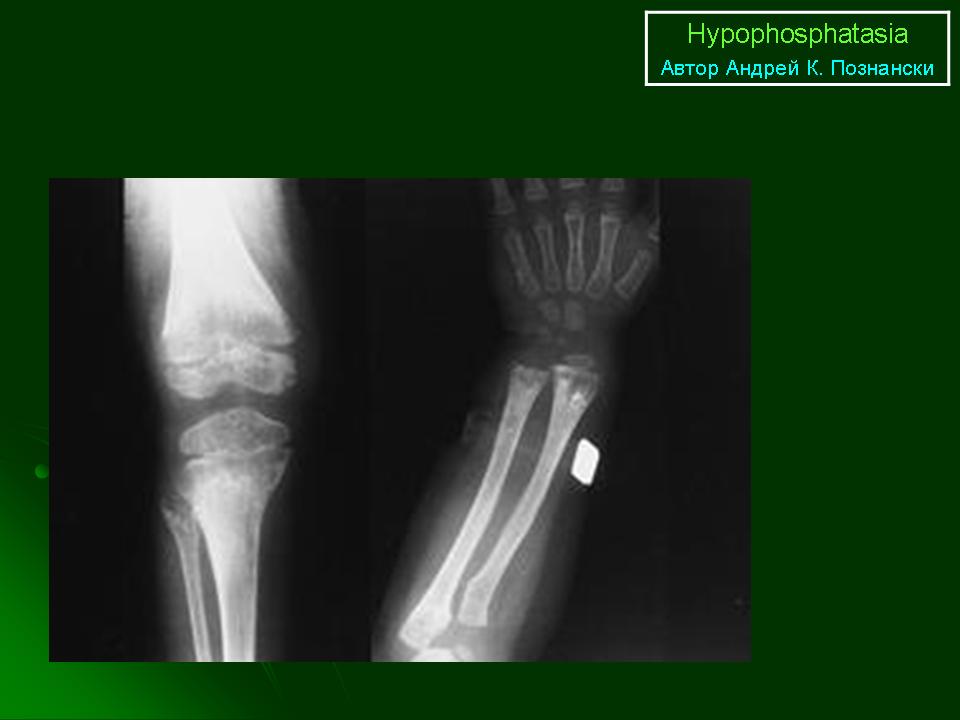 http://www.ncbi.nlm.nih.gov/pubmed/17519318
http://www.ncbi.nlm.nih.gov/pubmed/17519318
INTERNET
Mornet E, Nunes ME. Гипофосфатазия. 20 ноября 2007 г. [Обновлено 4 февраля 2016 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2021 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1150/ По состоянию на 26 января 2021 г.
Бакро С. и Морнет Э. Гипофосфатазия. сирота. Последнее обновление за февраль 2020 г. Доступно по адресу: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=162&Disease_Disease_Search_diseaseGroup=Hypophosphatasia&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Hypophosphasia&Hyposphatasia&title =Disease_Search_Simple По состоянию на 26 января 2021 г.
Correa Marquez RR и Behari G.. Гипофосфатазия. Медскейп. Последнее обновление: 7 августа 2019 г. Доступно по адресу: http://emedicine.medscape.com/article/945375-overview По состоянию на 26 января 2021 г.